As filed with the Securities and Exchange Commission on August 17, 2023.
Registration No. 333-272706
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
AMENDMENT NO. 1
TO
FORM F-10
REGISTRATION STATEMENT UNDER
THE SECURITIES ACT OF 1933
CYBIN INC.
(Exact name of Registrant as specified in its charter)
Ontario, Canada
(Province or other jurisdiction of incorporation or organization)
2834
(Primary Standard Industrial Classification Code Number, if applicable)
N/A
(I.R.S. Employer Identification No., if applicable)
100 King Street West, Suite 5600
Toronto, Ontario, Canada M5X 1C9
(908) 764-8385
(Address and telephone number of Registrant's principal executive offices)
C T Corporation System
1015 15th Street N.W., Suite 1000
Washington, DC 20005
(202) 572-3133
(Name, address (including zip code) and telephone number (including area code) of agent for service in the United States)
Copies to:
Douglas Drysdale
Cybin, Inc.
100 King Street West, Suite
5600
Toronto, Ontario, Canada
M5X 1C9
(866) 292-4601 |
Richard Raymer
Dorsey & Whitney LLP
TD Canada Trust Tower
Brookfield Place, 161 Bay
Street, Suite 4310 Toronto,
Ontario
Canada, M5J 2S1
(416) 367-7388 |
Approximate date of commencement of proposed sale of the securities to the public:
From time to time after this Registration Statement becomes effective.
Province of Ontario, Canada
(Principal jurisdiction regulating this offering)
It is proposed that this filing shall become effective (check appropriate box below):
| A. |
☒ |
upon filing with the Commission pursuant to Rule 467(a) (if in connection with an offering being made contemporaneously in the United States and Canada). |
| B. |
☐ |
at some future date (check the appropriate box below): |
| |
1. |
☐ |
|
pursuant to Rule 467(b) on ( ) at ( ) (designate a time not sooner than 7 calendar days after filing). |
| |
2. |
☐ |
|
pursuant to Rule 467(b) on ( ) at ( ) (designate a time 7 calendar days or sooner after filing) because the securities regulatory authority in the review jurisdiction has issued a receipt or notification of clearance on ( ). |
| |
3. |
☐ |
|
pursuant to Rule 467(b) as soon as practicable after notification of the Commission by the Registrant or the Canadian securities regulatory authority of the review jurisdiction that a receipt or notification of clearance has been issued with respect hereto. |
| |
4. |
☐ |
|
after the filing of the next amendment to this Form (if preliminary material is being filed). |
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to the home jurisdiction's shelf prospectus offering procedures, check the following box. ☒
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registration Statement shall become effective as provided in Rule 467 under the Securities Act or on such date as the Commission, acting pursuant to Section 8(a) of the Securities Act, may determine.
PART I
INFORMATION REQUIRED TO BE DELIVERED TO OFFEREES OR PURCHASERS
This short form base shelf prospectus has been filed under legislation in each of the provinces and the territories of Canada that permits certain information about these securities to be determined after this prospectus has become final and that permits the omission from this prospectus of that information. The legislation requires the delivery to purchasers of a prospectus supplement containing the omitted information within a specified period of time after agreeing to purchase any of these securities, except in cases where an exemption from such delivery requirements is available.
No securities regulatory authority has expressed an opinion about these securities and it is an offence to claim otherwise. This short form base shelf prospectus constitutes a public offering of these securities only in those jurisdictions where they may be lawfully offered for sale and therein only by persons permitted to sell such securities.
Information contained herein is subject to completion or amendment. A registration statement relating to these securities has been filed with the United States Securities and Exchange Commission (the "SEC"). These securities may not be sold nor may offers to buy be accepted prior to the time the registration statement becomes effective. This prospectus shall not constitute an offer to sell or the solicitation of an offer to buy nor shall be any sale of these securities in any state in which such offer, solicitation or sale would be unlawful prior to registration or qualification under the securities laws of any such state.
Information has been incorporated by reference in this short form base shelf prospectus from documents filed with securities commissions or similar authorities in Canada. Copies of the documents incorporated herein by reference may be obtained on request without charge from the Secretary of Cybin Inc. at 100 King Street West, Suite 5600, Toronto, Ontario M5X 1C9, telephone 1-866-292-4601, and are also available electronically at www.sedarplus.ca.
| New Issue |
August 17, 2023 |
SHORT FORM BASE SHELF PROSPECTUS

CYBIN INC.
$160,000,000
Common Shares
Warrants
Units
Debt Securities
Subscription Receipts
This short form base shelf prospectus ("Prospectus") relates to the offering for sale from time to time (each, an "Offering") by Cybin Inc. (the "Corporation" or "Cybin") during the 25-month period that this Prospectus, including any of amendments thereto, remains valid, of up to $160,000,000 in the aggregate of: (i) common shares ("Common Shares") of the Corporation; (ii) warrants ("Warrants") to purchase other Securities (as defined below) of the Corporation; (iii) units ("Units") comprising of one or more of the other Securities, (iv) senior and subordinated unsecured debt securities (collectively, "Debt Securities"), including debt securities convertible or exchangeable into other securities of the Corporation, and (v) subscription receipts ("Subscription Receipts" and together with the Common Shares, Warrants, Units and Debt Securities, collectively referred to herein as the "Securities"). The Securities may be offered separately or together, in amounts, at prices and on terms determined based on market conditions at the time of the sale and as set forth in an accompanying prospectus supplement ("Prospectus Supplement").
All shelf information permitted under applicable laws to be omitted from this Prospectus will be contained in one or more Prospectus Supplements that will be delivered to purchasers together with this Prospectus, except in cases where an exemption from such delivery requirements is available. Each Prospectus Supplement containing the specific terms of any Securities will be incorporated by reference into this Prospectus for the purposes of securities legislation as of the date of the Prospectus Supplement and only for the purposes of the distribution of the Securities to which the Prospectus Supplement pertains.
The specific terms of any Securities offered will be described in a Prospectus Supplement, including: (i) in the case of Common Shares, the number of Common Shares offered, the offering price (in the event the offering is a fixed price distribution), the manner of determining the offering price(s) (in the event the offering is a non-fixed price distribution) and any other specific terms; (ii) in the case of Warrants, the number of Warrants being offered, the offering price (in the event the offering is a fixed price distribution), the manner of determining the offering price(s) (in the event the offering is a non-fixed price distribution), the designation, number and terms of the other Securities purchasable upon exercise of the Warrants, and any procedures that will result in the adjustment of those numbers, the exercise price, the dates and periods of exercise and any other specific terms; (iii) in the case of Units, the number of Units offered, the offering price, the designation, number and terms of the other Securities comprising the Units, and any other specific terms; (iv) in the case of Debt Securities, the specific designation of the Debt Securities, whether such Debt Securities are senior or subordinate, the aggregate principal amount of the Debt Securities being offered, the currency or currency unit in which the Debt Securities may be purchased, authorized denominations, any limit on the aggregate principal amount of the Debt Securities of the series being offered, the issue and delivery date, the maturity date, the offering price (at par, at a discount or at a premium), the interest rate or method of determining the interest rate, the interest payment date(s), any conversion or exchange rights that are attached to the Debt Securities, any redemption provisions, any repayment provisions and any other specific terms; and (v) in the case of Subscription Receipts, the number of Subscription Receipts being offered, the offering price (in the event the offering is a fixed price distribution), the manner of determining the offering price(s) (in the event the offering is a non-fixed price distribution), the terms, conditions and procedures for the conversion of the Subscription Receipts into other Securities, the designation, number and terms of such other Securities, and any other specific terms. A Prospectus Supplement relating to a particular Offering of Securities may include terms pertaining to the Securities being offered thereunder that are not within the terms and parameters described in this Prospectus.
The Securities may be sold through underwriters or dealers, directly by the Corporation pursuant to applicable statutory exemptions, or through designated agents from time to time. See "Plan of Distribution". The Prospectus Supplement relating to a particular Offering of Securities will identify each underwriter, dealer or agent, as the case may be, engaged by the Corporation in connection with the offering and sale of the Securities, and will set forth the terms of the Offering of such Securities, including, to the extent applicable, any fees, discounts or any other compensation payable to underwriters, dealers or agents in connection with the Offering, the method of distribution of the Securities, the initial issue price (in the event that the offering is a fixed price distribution), the net proceeds to the Corporation and any other material terms of the plan of distribution.
The Securities may be sold from time to time in one or more transactions at a fixed price or prices or at non-fixed prices. This Prospectus may qualify an "at-the-market distribution", as defined in National Instrument 44-102 - Shelf Distributions ("NI 44-102"). If offered on a non-fixed price basis, the Securities may be offered at market prices prevailing at the time of sale, at prices determined by reference to the prevailing price of a specified security in a specified market or at prices to be negotiated with purchasers including sales in transactions that are deemed to be "at-the-market distributions", including sales made directly on the Neo Exchange Inc., now operating as Cboe Canada (the "NEO") or other existing trading markets for the Securities, and as set forth in an accompanying Prospectus Supplement, in which case the compensation payable to an underwriter, dealer or agent in connection with any such sale will be decreased by the amount, if any, by which the aggregate price paid for the Securities by the purchasers is less than the gross proceeds paid by the underwriter, dealer or agent to the Corporation. The price at which the Securities will be offered and sold may vary from purchaser to purchaser and during the period of distribution. See "Plan of Distribution".
This Prospectus does not qualify the issuance of Debt Securities in respect of which the payment of principal and/or interest may be determined, in whole or in part, by reference to one or more underlying interests including, for example, an equity or debt security, a statistical measure of economic or financial performance including, but not limited to, any currency, consumer price or mortgage index, or the price or value of one or more commodities, indices or other items, or any other item or formula, or any combination or basket of the foregoing items. For greater certainty, this Prospectus may qualify for issuance Debt Securities in respect of which the payment of principal and/or interest may be determined, in whole or in part, by reference to published rates of a central banking authority or one or more financial institutions, such as a prime rate or bankers' acceptance rate, or to recognized market benchmark interest rates such as the Canadian Overnight Repo Rate Average (CORRA), Secured Overnight Financing Rate (SOFR), Euro Inter-Bank Offered Rate (EURIBOR) or an alternative United States federal fund rate.
ii
No underwriter or dealer involved in an "at-the-market distribution" under this Prospectus, no affiliate of such an underwriter or dealer and no person or company acting jointly or in concert with such an underwriter or dealer will over-allot securities in connection with such distribution or effect any other transactions that are intended to stabilize or maintain the market price of the offered Securities or securities of the same class as the Securities distributed under the "at-the-market distribution", including selling an aggregate number or principal amount of Securities that would result in the underwriter creating an over-allocation position in the Securities.
In connection with any Offering of the Securities, subject to applicable laws and other than an "at-the-market distribution", the underwriters or agents may over-allot or effect transactions that stabilize or maintain the market price of the offered Securities at a level above that which might otherwise prevail on the open market. Such transactions, if commenced, may be interrupted or discontinued at any time. See "Plan of Distribution".
The Common Shares are listed on the NEO under the trading symbol “CYBN”, and in the United States on the NYSE American LLC stock exchange (the “NYSE American”) under the symbol “CYBN”. On August 16, 2023, the last trading day prior to the filing of this Prospectus, the closing prices of the Common Shares listed on the NEO and the NYSE American were $0.44 and US$0.323, respectively.
Unless specified in the applicable Prospectus Supplement, there is no market through which the Subscription Receipts, Warrants, Units and Debt Securities may be sold and purchasers may not be able to resell the Subscription Receipts, Warrants, Units and Debt Securities purchased under this Prospectus and the Prospectus Supplement. This may affect the pricing of the Subscription Receipts, Warrants, Units and Debt Securities in the secondary market, the transparency and availability of trading prices, the liquidity of the Subscription Receipts, Warrants, Units and Debt Securities and the extent of issuer regulation. See "Risk Factors".
The Corporation is a foreign private issuer under United States securities laws and is permitted under the multijurisdictional disclosure system adopted by the United States and Canada to prepare this Prospectus in accordance with Canadian disclosure requirements. Prospective investors should be aware that such requirements are different from those of the United States. The Corporation has prepared its financial statements, included or incorporated herein by reference, in accordance with International Financial Reporting Standards as issued by the International Accounting Standards Board and its consolidated financial statements are subject to Canadian generally accepted auditing standards and auditor independence standards. As a result, they may not be comparable to financial statements of United States companies.
Prospective investors should be aware that the purchase of Securities may have tax consequences that may not be fully described in this Prospectus or in any Prospectus Supplement, and should carefully review the tax discussion, if any, in the applicable Prospectus Supplement and in any event consult with a tax advisor.
An investors ability to enforce civil liabilities under the United States federal securities laws may be affected adversely because the Corporation is incorporated in Canada, most of the officers and directors named in this Prospectus are not residents of the United States, and some of the Corporation's assets and all or a substantial portion of the assets of such persons are located outside of the United States. See "Risk Factors - Enforcement of Civil Liabilities".
An investment in the Securities is subject to a number of risks, including those risks described in this Prospectus and documents incorporated by reference into this Prospectus. See “Risk Factors” in this Prospectus and in the Corporation’s Annual Information Form and Interim MD&A (each as defined herein) incorporated by reference herein.
iii
No person is authorized by the Corporation to provide any information or to make any representation other than as contained in this Prospectus in connection with the issue and sale of the Securities offered hereunder.
No underwriter has been involved in the preparation of this Prospectus or performed any review of the contents hereof.
NEITHER THE SEC OR ANY CANADIAN SECURITIES REGULATOR, NOR ANY STATE SECURITIES REGULATOR, HAS APPROVED OR DISAPPROVED THE SECURITIES OFFERED HEREBY OR PASSED UPON THE ACCURACY OR ADEQUACY OF THIS PROSPECTUS OR DETERMINED IF THIS PROSPECTUS IS TRUTHFUL OR COMPLETE. ANY REPRESENTATION TO THE CONTRARY IS A CRIMINAL OFFENCE.
Douglas Drysdale, an officer of the Corporation, resides outside of Canada. He has appointed Maxims CS Inc., Suite 1800, 181 Bay Street, Toronto, Ontario, M5J 2T9, as agent for service of process in Ontario. Prospective purchasers are advised that it may not be possible for investors to enforce judgments obtained in Canada against any person or company that is incorporated, continued or otherwise organized under the laws of a foreign jurisdiction or resides outside of Canada, even if the party has appointed an agent for service of process, see "Risk Factors - Risks Related to an Offering - Enforcement of Civil Liabilities".
In this Prospectus, references to the "Corporation", "Cybin", "we", "us" and "our" refer to Cybin Inc. and/or, as applicable, one or more of its subsidiaries. The Corporation's registered and head office is located at 100 King Street West, Suite 5600, Toronto, Ontario M5X 1C9.
|
The Corporation's current business focuses on conducting research and development of psychedelic therapeutics that aim to address unmet needs in the treatment of mental health conditions. This comprehensive development work is predicated on structural modifications of known tryptamine and phenethylamine derivatives to improve their pharmacokinetic properties while maintaining their respective pharmacology. Like most pharmaceutical companies, the Corporation's business is focused on research and development, see "Use of Proceeds".
The Canadian and United States federal governments regulate drugs through the Controlled Drugs and Substances Act (Canada) (the "CDSA") and the Controlled Substances Act (21 U.S.C. § 811) (the "CSA"), respectively, which place controlled substances in a schedule. Under the CDSA, psilocybin is currently a Schedule III drug. Under the CSA, psilocybin is currently a Schedule I drug.
In both Canada and the United States, the applicable federal government is responsible for regulating, among other things, the approval, import, sale and marketing of drugs, including any psychedelic substances, whether natural or novel. Health Canada, and the United States Food and Drug Administration ("FDA"), have not approved psilocybin as a drug for any indication. It is illegal to possess such substances without a prescription. The Corporation does not directly engage in any activities that would trigger the need to comply with any federal laws related to psychedelic substances. See "Regulatory Overview - Research and Development".
The Corporation does not deal with psychedelic substances except in jurisdictions where such activity is not illegal and then only within laboratory and clinical trial settings conducted within approved regulatory frameworks. The Corporation currently does not handle controlled or restricted substances under the CDSA or CSA. If the Corporation were to conduct this work without reliance on third parties, it would need to obtain the required licenses, approvals and authorizations from Health Canada, the FDA or other applicable regulatory bodies. The Corporation does not have any direct or indirect involvement with illegal selling, production or distribution of any substances in jurisdictions in which it operates.
|
iv
|
The Corporation's operations are conducted in strict compliance with local laws where such activities are permissible and do not require any specific legal or regulatory approvals.
Given the early stage of its prescription drug product development, the Corporation can make no assurance that its research and development program will result in regulatory approval or commercially viable products. To achieve profitable operations, the Corporation, alone or with others, must successfully develop, gain regulatory approval for, and market its future products. The Corporation currently has no products that have been approved by Health Canada, the FDA, or any similar regulatory authority. To obtain regulatory approvals for its prescription drug product candidates being developed and to achieve commercial success, clinical trials must demonstrate that the prescription drug product candidate are safe for human use and that they demonstrate efficacy. See "Risk Factors" herein and "Risk Factors" in the Annual Information Form.
Certain statements throughout this Prospectus regarding psilocybin, psychedelic tryptamine, tryptamine derivatives or other psychedelic compounds have not been evaluated by Health Canada, the FDA or other similar regulatory authorities, nor has the efficacy of psilocybin, psychedelic tryptamine, tryptamine derivatives or other psychedelic compounds been confirmed by approved research. There is no assurance that psilocybin, psychedelic tryptamine, tryptamine derivatives or other psychedelic compounds can be used to diagnose, treat, cure or prevent any disease or condition and robust scientific research and clinical trials are needed. There are multiple risk factors regarding the ability to successfully commercially scale a chemically synthesized process to obtain psilocybin and other analogues, including, but not limited to, regulatory changes or other changes in law, and risks related to drug development. See "Risk Factors" herein and "Risk Factors" in the Annual Information Form.
The Corporation oversees and monitors compliance with applicable laws in each jurisdiction in which it operates. In addition to the Corporation's senior executives and the employees responsible for overseeing compliance, the Corporation has local regulatory/compliance counsel engaged in every jurisdiction in which it operates. See "Compliance Program". Additionally, the Corporation has received legal opinions or advice in each jurisdiction where it currently operates regarding (a) compliance with applicable regulatory frameworks and (b) potential exposure and implications arising from applicable laws in jurisdictions where the Corporation has operations or intends to operate.
For these reasons, the Corporation may be (a) subject to heightened scrutiny by regulators, stock exchanges, clearing agencies and other authorities, (b) susceptible to regulatory changes or other changes in law, and (c) subject to risks related to drug development, among other things. There are a number of risks associated with the business of the Corporation. See "Risk Factors" herein and "Risk Factors" in the Annual Information Form.
An investment in the Securities is highly speculative and involves a high degree of risk that should be considered by potential purchasers. An investment in the Securities is suitable only for those purchasers who are willing to risk a loss of some or all of their investment and who can afford to lose some or all of their investment. A prospective purchaser should therefore review this Prospectus and the documents incorporated by reference herein in their entirety, including the Annual Information Form, and carefully consider the risk factors described under the section "Risk Factors" in this Prospectus, prior to investing in the Securities. See "Cautionary Note Regarding Forward-Looking Information" and "Risk Factors".
|
v
TABLE OF CONTENTS
Page
GENERAL MATTERS
Investors should rely only on the information contained in or incorporated by reference into this Prospectus or any applicable Prospectus Supplement. The Corporation has not authorized anyone to provide investors with different information. Information contained on the Corporation's website shall not be deemed to be a part of this Prospectus or incorporated by reference herein or in any applicable Prospectus Supplement and may not be relied upon by prospective investors for the purpose of determining whether to invest in the Securities qualified for distribution under this Prospectus. The Corporation is not making an offer of these Securities in any jurisdiction where the offer is not permitted. Investors should not assume that the information contained in this Prospectus is accurate as of any date other than the date on the front of this Prospectus or the date of the relevant document incorporated by reference. The Corporation's business, operating results, financial condition and prospects may have changed since that date.
The information contained on www.cybin.com is not intended to be included in or incorporated by reference herein, and prospective purchasers should not rely on such information when deciding whether or not to invest in any Securities.
In this Prospectus, unless stated otherwise or the context requires otherwise, all dollar amounts are expressed in Canadian dollars.
CAUTIONARY NOTE REGARDING FORWARD-LOOKING INFORMATION
Certain statements contained in this Prospectus, and in certain documents incorporated by reference herein, constitute "forward-looking information" and "forward-looking statements," within the meaning of applicable securities laws (collectively, "forward-looking statements"). All statements other than statements of historical fact, including, without limitation, those regarding the Corporation's future financial position, business strategy, budgets, research and development, plans and objectives of management for future operations, and any statements preceded by, followed by or that include the words "expect," "likely", "may," "will," "should," "intend," or "anticipate," "potential," "proposed," "estimate" and other similar words, including negative and grammatical variations thereof, or statements that certain events or conditions "may" or "will" happen, or by discussions of strategy, are forward-looking statements.
These statements are not historical facts but instead represent only the Corporation's expectations, estimates and projections regarding future events. These statements are not guarantees of future performance and involve assumptions, risks and uncertainties that are difficult to predict. Therefore, actual results may differ materially from what is expressed, implied or forecasted in such forward-looking statements. Additional factors that could cause actual results, performance or achievements to differ materially include, but are not limited to, those discussed under "Risk Factors" in the Annual Information Form and in this Prospectus and in other documents incorporated by reference in this Prospectus. Management provides forward-looking statements because it believes they provide useful information to readers when considering their investment objectives and cautions readers that the information may not be appropriate for other purposes. Consequently, all of the forward-looking statements made in this Prospectus and in documents incorporated by reference in this Prospectus are qualified by these cautionary statements and other cautionary statements or factors contained herein, and there can be no assurance that the actual results or developments will be realized or, even if substantially realized, that they will have the expected consequences to, or effects on, the Corporation. These forward-looking statements are made as of the date of this Prospectus and the Corporation assumes no obligation to update or revise them to reflect subsequent information, events or circumstances or otherwise, except as required by law.
The forward-looking statements in this Prospectus and in documents incorporated by reference in this Prospectus are based on numerous assumptions regarding the Corporation's present and future business strategies and the environment in which the Corporation will operate in the future, including assumptions regarding business and operating strategies, and the Corporation's ability to operate on a profitable basis.
Some of the risks which could affect future results and could cause results to differ materially from those expressed in the forward-looking statements contained herein include: the ongoing impact of the novel coronavirus COVID-19; limited operating history; achieving publicly announced milestones; speculative nature of investment risk; early stage of the industry and product development; regulatory risks and uncertainties; "foreign private issuer" status under the U.S. securities laws; plans for growth; limited products; limited marketing and sales capabilities; no assurance of commercial success; no profits or significant revenues; reliance on third parties for clinical development activities; risks related to third party relationships; reliance on contract manufacturers; safety and efficacy of products; clinical testing and commercializing products; completion of clinical trials; commercial grade product manufacturing; nature of regulatory approvals; unfavourable publicity or consumer perception; social media; biotechnology and pharmaceutical market competition; reliance on key executives and scientists; employee misconduct; business expansion and growth; negative results of external clinical trials or studies; product liability; enforcing contracts; product recalls; distribution and supply chain interruption; difficulty to forecast; promoting the brand; product viability; success of quality control systems; reliance on key inputs; liability arising from fraudulent or illegal activity; operating risk and insurance coverage; costs of operating as public company; management of growth; conflicts of interest; foreign operations; cybersecurity and privacy risk; environmental regulation and risks; decriminalisation of psychedelics; forward-looking statements may prove to be inaccurate; effects of inflation; political and economic conditions; application and interpretation of tax laws; enforcement of civil liabilities; Risks Related to Intellectual Property: trademark protection; trade secrets; patent law reform; patent litigation and intellectual property; protection of intellectual property; third-party licences; Financial and Accounting Risks: substantial number of authorized but unissued Common Shares; dilution; negative cash flow from operating activities; additional capital requirements; lack of significant product revenue; estimates or judgments relating to critical accounting policies; inadequate internal controls; Risks related to the Common Shares: market for the Common Shares; significant sales of Common Shares; volatile market price for the Common Shares; tax issues; no dividends; Risks related to an Offering and the Corporation: an investment in the Securities is highly speculative; completion of an Offering; receipt of all regulatory and stock exchange approvals in respect of an Offering; forward-looking statements may prove to be inaccurate; potential need for additional financing; negative operating cash flow and going concern; discretion over the use of proceeds; potential dilution; trading market; significant sales of Common Shares; positive return not guaranteed; management of growth; the Common Shares are subject to market price volatility; no history of payment of cash dividends; limited operating history as a public company; and risks relating to research and development objectives and milestones.
Although the forward-looking statements contained in this Prospectus are based upon what management currently believes to be reasonable assumptions, the Corporation cannot assure prospective investors that actual results, performance or achievements will be consistent with these forward-looking statements. In particular, the Corporation has made assumptions regarding, among other things:
• substantial fluctuation of losses from quarter to quarter and year to year due to numerous external risk factors, and anticipation that the Corporation will continue to incur significant losses in the future;
• uncertainty as to the Corporation's ability to raise additional funding to support operations;
• the Corporation's ability to access additional funding;
• the fluctuation of foreign exchange rates;
• the risks associated with the development of the Corporation's product candidates which are at early stages of development;
• reliance upon industry publications as the Corporation's primary sources for third-party industry data and forecasts;
• reliance on third parties to plan, conduct and monitor the Corporation's preclinical studies and clinical trials;
• reliance on third party contract manufacturers to deliver quality clinical and preclinical materials;
• the Corporation's product candidates may fail to demonstrate safety and efficacy to the satisfaction of regulatory authorities or may not otherwise produce positive results;
• risks related to filing investigational new drug applications to commence clinical trials and to continue clinical trials if approved;
• the risks of delays and inability to complete clinical trials due to difficulties enrolling patients;
• competition from other biotechnology and pharmaceutical companies;
• the Corporation's reliance on the capabilities and experience of the Corporation's key executives and scientists and the resulting loss of any of these individuals;
• the Corporation's ability to fully realize the benefits of acquisitions;
• the Corporation's ability to adequately protect the Corporation's intellectual property and trade secrets;
• the risk of patent-related or other litigation; and
• the risk of unforeseen changes to the laws or regulations in the United States, Canada, the United Kingdom, the Netherlands, Ireland and other jurisdictions in which the Corporation operates.
Drug development involves long lead times, is very expensive and involves many variables of uncertainty. Anticipated timelines regarding drug development are based on reasonable assumptions informed by current knowledge and information available to the Corporation. Every patient treated on future studies can change those assumptions either positively (to indicate a faster timeline to new drug applications and other approvals) or negatively (to indicate a slower timeline to new drug applications and other approvals). This Prospectus and the documents incorporated by reference herein contain certain forward-looking statements regarding anticipated or possible drug development timelines. Such statements are informed by, among other things, regulatory guidelines for developing a drug with safety studies, proof of concept studies, and pivotal studies for new drug application submission and approval, and assumes the success of implementation and results of such studies on timelines indicated as possible by such guidelines, other industry examples, and the Corporation's development efforts to date.
In addition to the factors set out above and those identified under the heading "Risk Factors" in the Annual Information Form and in this Prospectus, other factors not currently viewed as material could cause actual results to differ materially from those described in the forward-looking statements. Although the Corporation has attempted to identify important risks and factors that could cause actual actions, events or results to differ materially from those described in forward-looking statements, there may be other factors and risks that cause actions, events or results not to be anticipated, estimated or intended. Accordingly, readers should not place any undue reliance on forward-looking statements.
Many of these factors are beyond the Corporation's ability to control or predict. These factors are not intended to represent a complete list of the general or specific factors that may affect the Corporation. The Corporation may note additional factors elsewhere in this Prospectus and in any documents incorporated by reference into this Prospectus. All forward-looking statements speak only as of the date made. All subsequent written and oral forward-looking statements attributable to the Corporation, or persons acting on the Corporation's behalf, are expressly qualified in their entirety by the cautionary statements. Except as required by law, the Corporation undertakes no obligation to update any forward-looking statement.
The forward-looking statements contained in this Prospectus and the documents incorporated by reference herein are expressly qualified in their entirety by the foregoing cautionary statement. Investors should read this entire Prospectus, including the Annual Information Form, the documents incorporated by reference herein, and each applicable Prospectus Supplement, and consult their own professional advisers to ascertain and assess the income tax and legal risks and other aspects associated with holding Securities.
TRADEMARKS AND SERVICE MARKS
This Prospectus includes trademarks, trade names and service marks which are protected under applicable intellectual property laws for use in connection with the operation of the Corporation's business, and which are the property of the Corporation. All other trade names, trademarks or service marks appearing in this prospectus that are not identified as marks owned by the Corporation are the property of their respective owners. Solely for convenience, trademarks, service marks and trade names referred to in this prospectus may be listed without the ®, (TM) and (sm) symbols, however, we will assert, to the fullest extent under applicable law, the Corporation's applicable rights in these trademarks, service marks and trade names.
MARKETING MATERIALS
Any template version of marketing materials (as such terms are defined in National Instrument 41-101 - General Prospectus Requirements) that are utilized in connection with the distribution of Securities will be filed under the Corporation's profile on www.sedarplus.ca (SEDAR+). In the event that such marketing materials are filed after the date of the applicable Prospectus Supplement for the offering and before termination of the distribution of such Securities, such filed versions of the marketing materials will be deemed to be incorporated by reference into the applicable Prospectus Supplement for the purposes of the distribution of the Securities to which the Prospectus Supplement pertains.
MARKET AND INDUSTRY DATA
Market and industry data contained and incorporated by reference in this Prospectus or any applicable Prospectus Supplement concerning economic and industry trends is based upon good faith estimates of the Corporation's management or derived from information provided by industry sources. The Corporation believes that such market and industry data is accurate and that the sources from which it has been obtained are reliable. However, we cannot guarantee the accuracy of such information and we have not independently verified the assumptions upon which projections of future trends are based. While the Corporation is not aware of any misstatements regarding the industry data presented herein, the Corporation's estimates involve risks and uncertainties and are subject to change based on various factors, including those discussed under "Cautionary Note Regarding Forward-Looking Information" and "Risk Factors" in this Prospectus.
For the avoidance of doubt, nothing stated in this paragraph operates to relieve the Corporation or the underwriters/agents from liability for any misrepresentation in this Prospectus or under applicable Canadian securities laws.
DOCUMENTS INCORPORATED BY REFERENCE
Information has been incorporated by reference in this Prospectus from documents filed with securities commissions or similar authorities in Canada. Copies of the documents incorporated herein by reference may be obtained on request without charge from the Secretary of the Corporation at 100 King Street West, Suite 5600, Toronto, Ontario M5X 1C9, telephone (908) 764-8385, and are also available electronically on SEDAR+.
The following documents of the Corporation filed with the securities commissions or similar authorities in Canada are incorporated by reference in this Prospectus:
1. annual information form of the Corporation dated June 27, 2023 for the year ended March 31, 2023 (the “Annual Information Form”);
2. the audited consolidated financial statements of the Corporation and the notes thereto as at and for the fiscal year ended March 31, 2023, together with the auditor’s report thereon;
3. management’s discussion and analysis of the Corporation for the year ended March 31, 2023;
4. the unaudited interim condensed consolidated financial statements of the Corporation and the notes thereto for the three months ended June 30, 2023 (the “Interim Financial Statements”);
5. management’s discussion and analysis of the Corporation for the three months ended June 30, 2023 (the “Interim MD&A”);
6. management information circular of the Corporation dated July 13, 2022 relating to an annual meeting of shareholders of the Corporation held on August 15, 2022;
7. material change report dated May 31, 2023 relating to the sale of up to US$30,000,000 of Common Shares to Lincoln Park Capital Fund, LLC (the “Investor”), upon the terms and subject to the conditions contained in the purchase agreement dated May 30, 2023 (the “Purchase Agreement”), and made pursuant to and qualified by a prospectus supplement dated May 30, 2023 (the “May 2023 Prospectus Supplement”), to the Corporation’s short form base shelf prospectus dated July 5, 2021 (the “2021 Base Shelf Prospectus”); and
8. material change report dated August 8, 2023 relating to the public offering of 24,264,706 units of the Corporation (the “Units”) at a price of US$0.34 per Unit for gross proceeds of US$8,250,000 pursuant to a prospectus supplement dated August 1, 2023 to the 2021 Base Shelf Prospectus (the “Offering”).
Any document of the type referred to in Item 11.1 of Form 44-101F1 - Short Form Prospectus filed by the Corporation with a securities commission or similar regulatory authority in Canada after the date of this Prospectus and prior to 25 months from the date hereof shall be deemed to be incorporated by reference in this Prospectus.
In addition, to the extent any such document is included in any annual report on Form 40-F or 20-F (or any respective successor form) filed with the SEC subsequent to the date of this Prospectus and prior to 25 months from the date hereof, such document shall be deemed to be incorporated by reference as exhibits to the Registration Statement on Form F-10 (the "Registration Statement") of which this Prospectus forms a part. In addition, any other report on Form 6-K and the exhibits thereto filed or furnished by the Corporation with the SEC, and any other reports filed, under the United States Securities Exchange Act of 1934, as amended (the "Exchange Act") from the date of this Prospectus and prior to 25 months from the date hereof shall be deemed to be incorporated by reference as exhibits to the Registration Statement of which this Prospectus forms a part, but only if and to the extent expressly so provided in any such report.
Copies of the documents incorporated by reference herein will be available electronically on the Corporation's SEDAR+ profile, which can be accessed at www.sedarplus.ca, and from the Corporation's EDGAR profile at www.sec.gov. The Corporation's filings through SEDAR+ and EDGAR are not incorporated by reference in the Prospectus except as specifically set out herein.
Any statement contained in this Prospectus or in a document incorporated or deemed to be incorporated by reference in this Prospectus shall be deemed to be modified or superseded for the purposes of this Prospectus to the extent that a statement contained herein or in any subsequently filed document which also is or is deemed to be incorporated by reference in this Prospectus modifies or supersedes that statement. Any statement so modified or superseded shall not constitute a part of this Prospectus except as so modified or superseded. The modifying or superseding statement need not state that it has modified or superseded a prior statement or include any information set forth in the document that it modifies or supersedes. The making of a modifying or superseding statement shall not be deemed an admission for any purposes that the modified or superseded statement, when made, constituted a misrepresentation, an untrue statement of a material fact or an omission to state a material fact that is required to be stated or that is necessary to make a statement not misleading in light of the circumstances in which it was made.
Upon filing of a new annual information form and related annual financial statements with, and where required, accepted by, the applicable securities regulatory authorities during the currency of this Prospectus, the previous annual information form, including all amendments thereto, the previous annual financial statements and all interim financial statements (including any interim period management's discussion and analysis related thereto), material change reports and management information circulars filed prior to the commencement of the fiscal year in which the new annual information form is filed, shall be deemed no longer to be incorporated into this Prospectus for purposes of future offers and sales of Securities hereunder.
A Prospectus Supplement containing the specific terms of any Securities offered thereunder will be delivered to purchasers of such Securities together with this Prospectus to the extent required under applicable securities laws and will be deemed to be incorporated by reference into this Prospectus as of the date of such Prospectus Supplement solely for the purposes of the Securities offered hereunder and thereunder.
DOCUMENTS FILED AS PART OF THE REGISTRATION STATEMENT
The following documents have been, or will be, filed with the SEC as part of the Registration Statement of which this Prospectus forms a part: (i) the documents listed under "Documents Incorporated by Reference"; (ii) powers of attorney from certain of the Corporation's directors and officers (included on the signature page to the Registration Statement); (iii) the consent of Zeifmans LLP; and (iv) the form of indenture for any Debt Securities issued hereunder. A copy of any applicable form of warrant indenture, subscription receipt agreement or statement of eligibility of trustee on Form T-1, as applicable, will be filed by post-effective amendment or by incorporation by reference to documents filed or furnished with the SEC under the Exchange Act.
THE CORPORATION
This summary does not contain all the information that may be important to you in deciding whether to invest in the Securities. You should read the entire Prospectus, including the section entitled "Risk Factors", the applicable Prospectus Supplement, and the documents incorporated by reference herein, including the Annual Information Form, before making such decision.
Summary of the Business
The Corporation is a clinical-stage biopharmaceutical company on a mission to create safe and effective psychedelic-based therapeutics to address the unmet need for new and innovative treatment options for people who suffer from mental health conditions. The Corporation’s goal of revolutionizing mental healthcare is supported by a network of world-class partners and internationally recognized scientists aimed at progressing proprietary drug discovery platforms, innovative drug delivery systems, and novel formulation approaches and treatment regimens.1
The Corporation's research and development work focuses on a three-pillar strategy that leverages the Corporation's core competencies in preclinical innovation and clinical development. This strategy supports the creation of intellectual property ("IP") focused on developing the Corporation's platform technology, the progression of clinical development programs including CYB003, a deuterated psilocybin analog, CYB004, a deuterated version of N, N-dimethyltryptamine ("DMT"), CYB005, phenethylamine derivatives, and an expansive list of preclinical molecules to facilitate future drug development opportunities.
The Corporation currently has more than 50 pending patent applications across six patent families and spanning seven research programs through a combination of internal filings and external licensing agreements.
Advancement of Mental Healthcare
The Corporation is conducting research and development of psychedelic therapeutics that aim to address unmet needs in the treatment of mental health conditions. This comprehensive development work is predicated on structural modifications of known tryptamine and phenethylamine derivatives to improve their pharmacokinetic properties while maintaining their respective pharmacology.
Across its extensive research and development programs, the Corporation is evaluating a wide array of novel, synthetic psychedelic active pharmaceutical ingredients intended to be delivered through innovative drug delivery systems including via inhalation, via intravenous ("IV"), and intramuscular, or subcutaneous ("SC") administration.2
The Corporation intends to apply for regulatory approval for therapies targeting indications such as major depressive disorder, alcohol use disorder, generalized anxiety disorder and potentially other various mental health conditions3. The Corporation is also developing compounds that may have the potential to address neuroinflammation4, central nervous system disorders, and psychiatric disorders5.
For additional information in respect of the Corporation and its operation, please see the Corporation’s Annual Information Form and Interim MD&A incorporated by reference into this Prospectus.
_____________________________________________
1 This is a forward-looking statement that involves material assumptions by the Corporation. Drug development involves long lead times, is very expensive and involves many variables of uncertainty. Anticipated timelines regarding drug development are based on reasonable assumptions informed by current knowledge and information available to the Corporation. Such statements are informed by, among other things, regulatory guidelines for developing a drug with safety studies, proof of concept studies, and pivotal studies for new drug application submission and approval, and assumes the success of implementation and results of such studies on timelines indicated as possible by such guidelines, other industry examples, and the Corporation's development efforts to date.
2 See footnote 1.
3 See footnote 1.
Inter-Corporate Relationships
As at the date of this Prospectus, the Corporation's corporate structure includes the following material wholly-owned subsidiaries:
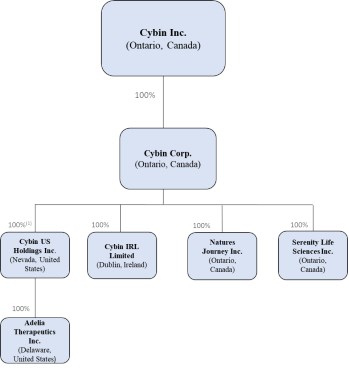
Note:
(1) The shareholders of Adelia Therapeutics Inc. ("Adelia") hold certain non-voting securities of Cybin U.S. issued in connection with the acquisition of Adelia on December 14, 2020 (the "Adelia Transaction"). For additional information in respect of the Adelia Transaction, please see the Annual Information Form incorporated by reference in this Prospectus Supplement.
Select Recent Developments
Other than as set forth below and generally in this Prospectus or the documents incorporated by reference herein, there have been no material developments in the business of the Corporation since August 14, 2023, the date of the Interim Financial Statements and Interim MD&A.
On May 30, 2023, the Corporation announced it had entered into the Purchase Agreement with the Investor, pursuant to which the Investor agreed to purchase from the Corporation, at the Corporation’s direction from time to time, in the Corporation’s sole discretion, from and after the date that the conditions to the Investor’s purchase obligations set forth in the Purchase Agreement are satisfied, including the filing of the May 2023 Prospectus Supplement, and over a period of up to 36 months thereafter, Common Shares having a total maximum aggregate purchase price to the Investor of US$30,000,000 (subject to certain limitations contained in the Purchase Agreement), upon the terms and subject to the conditions contained in the Purchase Agreement. On July 31, 2023, the Corporation issued a notice of suspension of the Purchase Agreement to the Investor in the context of announcing the Offering. The Corporation sold a total of US$465,273 under the Purchase Agreement out of the total US$30,000,000 qualified under the May 2023 Prospectus Supplement, using the proceeds for the advancement of its research and development programs and for general corporate purposes. The Corporation has suspended all sales under the Purchase Agreement allowing it to use the balance of US$29,534,727 available under the 2021 Base Shelf Prospectus for the Offering. Subject to receipt of all necessary regulatory approvals, the Corporation intends to file a prospectus supplement under this Prospectus with respect to the Purchase Agreement.
On June 29, 2023, the Corporation announced the appointment of Aaron Bartlone as Chief Operating Officer, effective July 1, 2023. Mr. Bartlone has served as Chief Operating Officer of Cybin’s U.S. subsidiary, Cybin US Holdings, Inc., since March 2021. Mr. Bartlone is a United States resident and currently holds 701,070 Common Shares and options exercisable into 1,315,000 Common Shares.
On August 5, 2023, the Corporation’s at-the-market equity program to issue and sell up to US$35,000,000 of Common Shares (the “ATM Program”) automatically terminated in accordance with the terms of the equity distribution agreement entered into on August 8, 2022, due to the Corporation’s prior base shelf prospectus, dated July 5, 2021, lapsing on August 5, 2023, being 25 months after the effective date. Subject to receipt of all necessary approvals and the entering into of a new equity distribution agreement, the Corporation intends to file a prospectus supplement under this Prospectus in order to qualify the issuance and sale of Common Shares pursuant to an at-the-market equity program.
On August 15, 2023, the Corporation announced that the U.S. Patent and Trademark Office has granted U.S. patent 11,724,985, to a deuterated psilocybin analog in the Corporation’s CYB003, investigational drug program. The patent, which is expected to provide exclusivity until 2041, includes composition of matter claims to deuterated tryptamines in support of the Corporation’s clinical-stage programs, CYB003, a proprietary deuterated psilocybin analog, and CYB004, a proprietary deuterated dimethyltryptamine, in addition to other of the Corporation’s pre-clinical programs, as well as claims directed towards methods of treating major depressive disorder (“MDD”) and treatment-resistant depression.
On August 17, 2023, the Corporation announced that it has initiated preparations for good manufacturing practices (“GMP”) production of a capsule formulation of CYB003, its proprietary deuterated psilocybin analog in development for the potential treatment of MDD.
REGULATORY OVERVIEW
A summary of the applicable regulatory framework for the Corporation's various business segments and proposed business activity are set forth below.
Canada
In Canada, oversight of healthcare is divided between the federal and provincial governments. The federal government is responsible for regulating, among other things, the approval, import, sale, and marketing of drugs such as psilocybin and other psychedelic substances, whether natural or novel. The provincial/territorial level of government has authority over the delivery of health care services, including regulating health facilities, administering health insurance plans such as the Ontario Health Insurance Plan, distributing prescription drugs within the province, and regulating health professionals such as doctors, psychologists, psychotherapists and nurse practitioners. Regulation is generally overseen by various colleges formed for that purpose, such as the College of Physicians and Surgeons of Ontario.
Certain psychoactive compounds, such as psilocybin, are considered controlled substances under Schedule III of the CDSA. In order to conduct any scientific research, including pre-clinical and clinical trials, using psychoactive compounds listed as controlled substances under the CDSA, an exemption under Section 56 of the CDSA (a "Section 56 Exemption") is required. This exemption allows the holder to possess and use the controlled substance without being subject to the restrictions set out in the CDSA.
Health Canada has not approved psilocybin as a drug for any indication. However, there are legal routes through which psilocybin may be accessed for medical or scientific purposes. The Canadian Minister of Health can grant Section 56 Exemptions if it is deemed to be necessary for a medical or scientific purpose or is otherwise in the public interest. The Corporation has not applied for a Section 56 Exemption from Health Canada.
Health Canada's Special Access Program ("SAP") was designed to provide Canadians to access certain restricted drugs before they are formally approved for use in Canada. In January 2022, certain amendments to the SAP came into force to permit medical practitioners treating patients with serious or life-threatening conditions to request access to restricted drugs that have not yet been approved for sale in Canada when conventional therapies have failed, are unsuitable, or unavailable in Canada. Such amendments create a means of legally accessing psilocybin through the SAP. The Corporation has not applied for access under the SAP.
The possession, sale or distribution of controlled substances is prohibited unless specifically permitted by the government. A party may seek government approval for a Section 56 Exemption to allow for the possession, transport or production of a controlled substance for medical or scientific purposes. Products that contain a controlled substance such as psilocybin cannot be made, transported or sold without proper authorization from the government. A party can apply for a Dealer's License under the Food and Drug Regulations (Part J). In order to qualify as a licensed dealer, a party must meet all regulatory requirements mandated by the regulations including having compliant facilities, compliant materials and staff that meet the qualifications under the regulations of a senior person in charge and a qualified person in charge. Assuming compliance with all relevant laws (CDSA, Food and Drugs Regulations) and subject to any restrictions placed on the license by Health Canada, an entity with a Dealer's License may produce, assemble, sell, provide, transport, send, deliver, import or export a restricted drug (as listed in Part J in the Food and Drugs Regulations - which includes psilocybin and psilocin) (see s. J.01.009 (1) of the Food and Drug Regulations).
The Corporation intends to sponsor and work with licensed third parties to conduct any clinical trials and research and does not handle controlled substances. If the Corporation were to conduct this work without the reliance on third parties, it would need to obtain additional licenses and approvals described above.
Please see "Description of the Business - Research and Development" in the Annual Information Form for additional information concerning the regulation applicable to the process required before prescription drug product candidates may be marketed in Canada.
United States
The FDA and other federal, state, local and foreign regulatory agencies impose substantial requirements upon the clinical development, approval, labeling, manufacture, marketing and distribution of drug products. These agencies regulate, among other things, research and development activities and the testing, approval, manufacture, quality control, safety, effectiveness, labeling, storage, record keeping, advertising and promotion of any prescription drug product candidates or commercial products. The regulatory approval process is generally lengthy and expensive, with no guarantee of a positive result. Moreover, failure to comply with applicable FDA or other requirements may result in civil or criminal penalties, recall or seizure of products, injunctive relief including partial or total suspension of production, or withdrawal of a product from the market. The Corporation intends to file an investigational new drug application (“IND”) related to its Deuterated Psilocybin Analog Program upon completion of its pre-clinical studies and CMC development.6 Anticipated timelines related to regulatory filings are based on reasonable assumptions informed by current knowledge and information available to the Corporation.
Psilocybin, psilocin, dimethyltryptamine, and 5-Methoxy-N-N-dimethyltryptamine are strictly controlled under the CSA as Schedule I substances. Schedule I substances by definition have no currently accepted medical use in the United States, a lack of accepted safety for use under medical supervision, and a high potential for abuse. Schedule I and II drugs are subject to the strictest controls under the CSA, including manufacturing and procurement quotas, security requirements and criteria for importation. Anyone wishing to conduct research on substances listed in Schedule I under the CSA must register with the United States Drug Enforcement Administration ("DEA") and obtain DEA approval of the research proposal. A majority of state laws in the United States also classify psilocybin and psilocin as Schedule I controlled substances. For any product containing psilocybin or any Schedule I substance to be available for commercial marketing in the United States, such substance must be rescheduled, or the product itself must be scheduled, by the DEA to Schedule II, III, IV or V. Scheduling determinations by the DEA are dependent on FDA approval of a substance or a specific formulation of a substance.
See "Description of the Business - Research and Development" in the Annual Information Form for additional information concerning the regulation applicable to the process required before prescription drug product candidates may be marketed in the United States.
Europe (Netherlands)
The International Narcotics Control Board ("INCB"), a United Nations ("UN") entity, monitors enforcement of restrictions on controlled substances. The INCB's authority is defined by three international UN treaties - the UN Single Convention on Narcotic Drugs of 1961, the UN Psychotropic Convention of 1971 (referred to herein as the UN71), and the UN Convention Against Illicit Traffic in Narcotic Drugs and Psychotropic Substances of 1988, which contains provisions related to the control of controlled substance precursors. EU Member States, including the Netherlands, that have agreed to abide by the provisions of these treaties, each create responsible agencies and enact laws or regulations to implement the requirements of these conventions.
Specific EU legislation establishing different classes of controlled substances is limited to EU regulations that define classes of precursors, or substances used in the illicit manufacture of controlled substances, including Regulation (EC) No. 273/2004 of the European Parliament and the Council of February 11, 2004 and the Council Regulation (EC) No. 111/2005 of December 22, 2004. While EU legislation does not establish different classes of narcotic drugs or psychotropic substances, the Council Decision 2005/387/JHA of May 10, 2005 can provoke a Council Decision requiring EU member states to put a drug under national controls equivalent to those of the INCB. DMT is currently classified as a Schedule I substance under the UN71; the EU member states that are party to the UN71, including the Netherlands, have agreed to the following in respect of Schedule I substances:
_____________________________________________
6 This statement is based on the following material assumption: drug development involves long lead times, is very expensive and involves many variables of uncertainty. Anticipated timelines regarding drug development are based on reasonable assumptions informed by current knowledge and information available to the Corporation. As of the date hereof, it has not yet completed the aforementioned items. Such statements are informed by, among other things, regulatory guidelines for developing a drug with safety studies, proof of concept studies, and pivotal studies for new drug application submission and approval, and assumes the success of implementation and results of such studies on timelines indicated as possible by such guidelines, other industry examples, and the Corporation’s development efforts to date. See “Risk Factors”.
- prohibit all use except for scientific and very limited medical purposes by duly authorized persons, in medical or scientific establishments which are directly under the control of their Governments or specifically approved by them;
- require that manufacture, trade, distribution and possession be under a special licence or prior authorization;
- provide for close supervision of the activities and acts mentioned in paragraphs (a) and (b);
- restrict the amount supplied to a duly authorized person to the quantity required for his authorized purpose;
- require that persons performing medical or scientific functions keep records concerning the acquisition of the substances and the details of their use, such records to be preserved for at least two years after the last use recorded therein; and
- prohibit export and import except when both the exporter and importer are the competent authorities or agencies of the exporting and importing country or region, respectively, or other persons or enterprises which are specifically authorized by the competent authorities of their country or region for the purpose.
As classification of controlled substances may vary among different EU member states, sponsors must be aware of the prevailing legislation in each country where a clinical trial may be conducted. Prior to operating or conducting any preclinical or clinical studies in any other EU member state, Cybin will investigate the specific regulatory requirements of such EU member state. As referenced above, a licence is required for individuals and entities who wish to produce, dispense, import, or export Schedule I substances (including DMT), but the specific requirements vary from country to country. Currently, DMT is classified in the Netherlands as a List 1 Drug under the Dutch Opium Act (Opiumwet) (the "Dutch Opium Act") and as such, subject to express authorization being obtained, the production, trade and possession of DMT are prohibited.
In addition to the Dutch Opium Act, two other Dutch Acts may be relevant when it comes to drugs: the Medicines Act and the Commodities Act.
The specific regulatory processes and approvals required may vary among different EU member states and are set forth in the respective legislation of each country. For The Netherlands, there are specific regulatory requirements for the approval of clinical trials that need to be met. Firstly, a CTA (Clinical Trial Application) dossier containing the preclinical and any clinical information along with the proposed clinical trial design must be submitted to an accredited Ethics Committee and to the Central Commission on Research in Humans (the "CCMO"), which is also known as the Competent Authority in The Netherlands. In Dutch, the CCMO is called the 'Centrale Commissie Mensgebonden Onderzoek'. In cases where the study involves a substance subject to the Dutch Opium Act (such as DMT), an official exemption by Farmatec is needed, which needs to be included in the CTA.
Specific rules for the submission, assessment and conduct of clinical trials with medicinal products are set out in, among others, the EU Clinical Trial Regulation 536/2014 (CTR), which is applicable in the EU as of January 31, 2022 and the Medical Research (Human Subjects) Act (Wet medisch-wetenschappelijk onderzoek met mensen).
Ireland
In Ireland, psilocin is a controlled substance under the Misuse of Drugs Act, 1977, 1984 and 2015 (the "Ireland MDA"), the Misuse of Drugs Regulations 2017 (the "Ireland MDR") and the Criminal Justice (Psychoactive Substances) Act 2010. These are the primary legislative instruments which govern controlled substances in Ireland. This legislation regulates the use, possession, supply, licensing, and administration of listed scheduled substances and establishes the offences and penalties for anything done contrary to the legislation. Any substance, product or preparation (whether natural or otherwise) including a fungus of any kind or description, which contains psilocin or an ester of psilocin is controlled as a Schedule 1 controlled substance under the Ireland MDA and the Ireland MDR. The Ireland MDR includes "any substance, product or preparation including fungi of any kind or description, containing psilocin or an ester of psilocin (which are commonly described as 'magic mushrooms')" within the strict regime of control that applies to those substances in Schedule 1 of the Ireland MDR. Accordingly, psilocin will qualify as a Schedule 1 controlled substance and is subject to the strict regime of control that applies. As a Schedule 1 controlled substance under the Ireland MDA, unlawful manufacturing, production, preparation, importation, exportation, supply, or distribution of psilocin carries onerous obligations and harsh punishments for contravention; this include fines and/or terms of imprisonment of up to 14 years. Pursuant to the Ireland MDA, in certain circumstances, the Minister for Health "may grant licences or issue permits or authorizations for any of the purposes of this Act, attach conditions to any such licence, permit or authorization, vary such conditions and revoke any such licence, permit or authorization". Where licences are granted, there are very strict conditions imposed on licence holders. For example, strict conditions can be placed regarding the security, storage and documenting controlled substances.
The Corporation does not currently engage in any activities in Ireland that are regulated by such laws. If the Corporation were to engage in such activities, it would need to obtain the appropriate licences and authorization to do so. The Corporation intends to constantly review its Irish operations to ensure compliance with all applicable laws as the operations evolve.
United Kingdom
In the United Kingdom, there are two main "layers" of regulation with which products containing controlled substances must comply. These are: (i) controlled drugs legislation, which applies to all products irrespective of the type of product, and (ii) the regulatory framework applicable to a specific category of products, in this case, pharmaceuticals and food/food supplements.
The main controlled drugs legislation in the United Kingdom is the Misuse of Drugs Act 1971 ("MDA") and the Misuse of Drugs Regulations 2001 ("MDR"), each as amended. The MDA sets out the penalties for unlawful production, possession and supply of controlled drugs based on three classes of risk (A, B and C). The MDR sets out the permitted uses of controlled drugs based on which Schedule (1 to 5) they fall within.
In the United Kingdom, "Fungus (of any kind) which contains psilocin or an ester of psilocin" is controlled as a Class A drug under the MDA and Schedule 1 drug under the MDR. As psilocybin is a phosphate ester of psilocin, even if it is isolated from psilocin, it will still be treated as a Class A drug under the MDA and as a Schedule 1 drug under the MDR.
In the United Kingdom, Class A drugs are deemed to be the most dangerous, and so carry the harshest punishments for unlawful manufacture, production, possession and supply. Schedule 1 drugs can only be lawfully manufactured, produced, possessed and supplied under a controlled drugs domestic licence issued by the UK Home Office. While exemptions do exist, none are applicable to the API.
The Corporation previously disclosed that it intended to file a clinical trial application with the United Kingdom Medicines and Healthcare products Regulatory Agency (the "MHRA") related to the Deuterated Psilocybin Analog Program upon completion of its pre-clinical studies and CMC development. The Corporation has since decided that it will first proceed in the United States and will re-evaluate other applications at a later date. Anticipated timelines related to regulatory filings are based on reasonable assumptions informed by current knowledge and information available to the Corporation.
Licensing Requirements
The Corporation obtains CYB003 API from a pharmaceutical ingredient provider who is FDA-registered and based in the United States. The API itself has been manufactured and packaged in FDA-registered facilities in the United States. The API is expected to be sent directly to the Corporation's partners for research and development purposes in the United States, Canada and the United Kingdom and to its clinical trial site in the U.S. As a part of the Corporation's acquisition of a Phase 1 DMT study from Entheon Biomedical Corp. in July 2022 (the "Asset Acquisition"), the Corporation also acquired API. The CYB004-E API was manufactured in the Netherlands by a pharmaceutical ingredient provider that is United States FDA-inspected.7
_____________________________________________
7 As a result of the Asset Acquisition, including the existing API, the Corporation did not direct the manufacturing of the API for CYB004-E and proceeded in reliance upon the representations of Entheon Biomedical Corp. and the Corporation's acquisition diligence. While the Corporation believes the CYB004- E API meets all required specifications, the Corporation did not oversee or direct the manufacture of the DMT API being used in CYB004-E.
Although the facilities in the UK are currently FDA-registered, this would not be sufficient to ensure the existence of valid marketing activities at this site. As mentioned above, in order to produce, possess and supply the API, the UK-based facility must also hold a domestic licence issued by the Home Office covering the manufacture, production, possession and supply of a controlled substance, as well as an export licence for each API shipment. The export application must include details of the importer and any import licence required by the local authorities in the United States. Moreover, as set out below in more detail under the heading "Pharmaceutical Products", depending on how the API is developed, certain authorizations and licences from the MHRA may be required to authorize some of the activities carried on at the UK-based facilities in relation to the API.
All premises that are licensed in connection with the possession, supply, manufacture and/or production of controlled drugs are required to adhere to detailed security standards.8
Typically, when controlled drugs are being transported between licensees, responsibility for their security remains with the owner and does not transfer to either the courier or the customer until the drugs arrive at their destination and are signed for. However, where a third party is involved in the transit and/or storage of controlled drugs, even if they are not the legal owners, this party also carries responsibility for their security by virtue of being 'in possession' of them. Under the Home Office guidance, each organisation involved in the movement of controlled drugs should have a standard operating procedure covering their responsibilities, record keeping, reconciliation and reporting of thefts/losses.9
Pharmaceutical Products
A product is regulated as a "medicinal product" under UK legislation (the Human Medicines Regulations 2012) if (i) it is a substance or combination of substances presented as having properties of preventing or treating disease in human beings (e.g., in marketing claims) or (ii) it is a substance or combination of substances that may be used by or administered to human beings with a view to (a) restoring, correcting or modifying a physiological function by exerting a pharmacological, immunological or metabolic action, or (b) making a medical diagnosis.
Whether a specific product restores, corrects or modifies a physiological function by exerting a pharmacological, immunological or metabolic action will depend on factors such as the concentration of the psilocybin/psilocin and the mode of action of any psilocybin/psilocin absorbed in the body.
If a product is a medicinal product, a marketing authorization for the product is required before the product can be placed on the market in the UK. The process for obtaining a marketing authorization involves submitting preclinical and clinical data as well as quality and manufacturing information in the form of a common technical document. In addition to a marketing authorization for the product itself, companies carrying out activities involving medicinal products, such as manufacturing, distribution and wholesaling, need to meet defined standards GMP and/or Good Distribution Practice ("GDP")) and to hold a related licence from the MHRA
How the API is subsequently processed will determine the licences that the UK-based facility must hold. In particular:
-
if the API is just one 'ingredient' of the investigational medicinal product ("IMP") which is used in the clinical trial then the UK-based facility must register with the MHRA and provide the MHRA with 60 days' notice of the intended start of manufacture/distribution, and comply with GMP and GDP for active substances; and
-
conversely, if the API will itself constitute the IMP, the manufacturer must, except in certain limited circumstances, hold a Manufacturer's Authorisations for IMPs licence ("MIA(IMP)"). In this scenario, an MIA(IMP) would be required regardless of whether the IMP is for use in the UK, another EEA Member State or a third country (such as the United States or Canada).
_____________________________________________
8 Home Office guidance; Security guidance for all existing or prospective Home Office Controlled Drug Licensees and/or Precursor Chemical Licensees or Registrants; 2022; https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1125889/ Security_Guidance_for_all_Businesses_and_Other_Organisations_v1.5_Nov_2022.pdf.
9 Home Office guidance; Guidelines for Standard Operating Procedures (SOPs); https://assets.publishing.service.gov.uk/government/uploads/ system/uploads/attachment_data/file/480572/StandardOpProcedure.pdf.
Some products fall on the borderline between medicines and another category such as medical devices, cosmetics or food supplements. The regulatory status of the product will be determined by i) the actual effect of the product on the body; and ii) any claims made about the effect of the product. Where a product is potentially both a medicinal product and another category of product, the legal position in the UK and EU is that it will be regulated as a medicinal product.
Research and Development
The Corporation is focused on development of psychedelic medicines and other products, through research and development of novel chemical compounds and delivery mechanisms and study of such compounds in clinical environments around the world. The Corporation anticipates growing its pipeline of psychedelic pharmaceutical products through its internal research, development, proprietary discovery programs, mergers and acquisitions, joint ventures and collaborative development agreements. For the time being, the Corporation maintains intellectual property generated by its R&D programs through patent filings and as trade secrets. The Corporation anticipates that as these programs mature more patent applications will be filed and more details about these programs will be disclosed at such time.
As a result of COVID-19, certain institutions have implemented certain facility procedures and are utilizing technology in an effort to mitigate the effects of the pandemic, specifically by moving patient interactions to remote status wherever possible. The Corporation cannot guarantee that the continued effects of COVID-19 will not impact patient recruiting for clinical trials and institutional processes at institutions involved in pharmaceutical product development.
Psychedelics are a class of drug whose primary action is to trigger psychedelic experiences by way of serotonin receptor agonism, causing thought, visual and auditory changes, and altered state of consciousness. Major psychedelic drugs include mescaline, LSD, psilocybin, and DMT. Psilocybin is a naturally occurring psychedelic prodrug compound produced by more than 200 species of mushrooms, collectively known as psilocybin mushrooms. The most potent are members of the genus Psilocybe, such as P. azurescens, P. semilanceata, and P. cyanescens, but psilocybin has also been isolated from about a dozen other genera. As a prodrug, psilocybin is quickly converted by the body to psilocin, which has mind-altering effects.
The pharmacokinetics, pharmacology and human metabolism of psilocybin are well known and well characterized. In conjunction with psychotherapy, psilocybin has been utilized broadly in phase II clinical trials.
Psilocybin found in certain species of mushrooms is a non-habit forming naturally occurring psychedelic compound. Once ingested, psilocybin is rapidly metabolized to psilocin, which then acts on serotonin receptors in the brain.
Cybin has commenced research and development on the delivery of synthetic psilocybin and other psychedelics through mechanisms such as sublingual film delivery, IV, and by way of inhalation.
The Corporation's research and development must be conducted in strict compliance with the regulations of federal, state, local and regulatory agencies in Canada, the United States, and the United Kingdom and the equivalent regulatory agencies in the other jurisdictions in which the Corporation operates. These regulatory authorities regulate, among other things, the research, manufacture, promotion and distribution of drugs in specific jurisdictions under applicable laws and regulations.
Canada
The process required before a prescription drug product candidate may be marketed in Canada generally involves:
- Chemical and Biological Research - Laboratory tests are carried out on tissue cultures and with a variety of small animals to determine the effects of the drug. If the results are promising, the manufacturer will proceed to the next step of development.
-
Pre-Clinical Development - Animals are given the drug in varying amounts over differing periods of time. If it can be shown that the drug causes no serious or unexpected harm at the doses required to have an effect, the manufacturer will proceed to clinical trials.
-
Clinical Trials - Phase I - The first administration in humans is to test if people can tolerate the drug. If this testing is to take place in Canada, the manufacturer must prepare a clinical trial application for the Therapeutic Products Directorate of Health Canada (the "TPD"). This includes the results of the first two steps and a proposal for testing in humans. If the information is sufficient, the Health Products and Food Branch of Health Canada (the "HPFB") grants permission to start testing the drug, generally first on healthy volunteers.
-
Clinical Trials - Phase II - Phase II trials are carried out on people with the target condition, who are usually otherwise healthy, with no other medical condition. Trials carried out in Canada must be approved by the TPD. In phase II, the objective of the trials is to continue to gather information on the safety of the drug and begin to determine its effectiveness.
-
Clinical Trials - Phase III - If the results from phase II show promise, the manufacturer provides an updated clinical trial application to the TPD for phase III trials. The objectives of phase III include determining whether the drug can be shown to be effective, and have an acceptable side effect profile, in people who better represent the general population. Further information will also be obtained on how the drug should be used, the optimal dosage regimen and the possible side effects.
-
New Drug Submission - If the results from phase III continue to be favourable, the drug manufacturer can submit a new drug submission ("NDS") to the TPD. A drug manufacturer can submit an NDS regardless of whether the clinical trials were carried out in Canada. The TPD reviews all the information gathered during the development of the drug and assesses the risks and benefits of the drug. If it is judged that, for a specific patient population and specific conditions of use, the benefits of the drug outweigh the known risks, the HPFB will approve the drug by issuing a notice of compliance.
United States
Because psilocybin, psilocin, DMT, and 5-Methoxy-DMT are listed as Schedule I substances under the CSA, for any product containing psilocybin or any Schedule I substance to be available for commercial marketing in the United States, such substance must be rescheduled, or the product itself must be scheduled, by the DEA to Schedule II, III, IV or V.
The process required before a prescription drug product candidate may be marketed in the United States generally involves:
-
completion of extensive non-clinical laboratory tests, animal studies and formulation studies, all performed in accordance with the FDA's Good Laboratory, Good Clinical and/or Manufacturing Practice regulations;
-
submission to the FDA of an IND, which must become effective before human clinical trials may begin;
-
approval by an institutional review board or independent ethics committee at each clinical trial site before each trial may be initiated;
-
for some products, performance of adequate and well-controlled human clinical trials in accordance with the FDA's regulations, including Good Clinical Practices, to establish the safety and efficacy of the prescription drug product candidate for each proposed indication;
-
submission to the FDA of a New Drug Application ("NDA"); and
-
FDA review and approval of the NDA prior to any commercial marketing, sale or shipment of the drug.
The testing and approval process requires substantial time, effort and financial resources, and the Corporation cannot be certain that the DEA will schedule or reschedule any Schedule I substance or product candidate to Schedule II, III, IV, or V, or that approvals for its prescription drug product candidates will be granted on a timely basis, if at all.
Non-clinical tests include laboratory evaluations of product chemistry, formulation and stability, as well as studies to evaluate toxicity in animals and other animal studies. The results of non-clinical tests, together with manufacturing information and analytical data, are submitted as part of an IND to the FDA. Some non-clinical testing may continue even after an IND is submitted. The IND also includes one or more protocols for the initial clinical trial or trials and an investigator's brochure. An IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or questions relating to the proposed clinical trials as outlined in the IND and places the clinical trial on a clinical hold. In such cases, the IND sponsor and the FDA must resolve any outstanding concerns or questions before any clinical trials can begin. Clinical trial holds also may be imposed at any time before or during studies due to safety concerns or non-compliance with regulatory requirements.
An independent institutional review board ("IRB"), at each of the clinical centers proposing to conduct the clinical trial must review and approve the plan for any clinical trial before it commences at that center. An IRB considers, among other things, whether the risks to individuals participating in the trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the consent form signed by the trial participants and must monitor the study until completed. The FDA, the IRB, or the sponsor may suspend or discontinue a clinical trial at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk. There also are requirements governing the reporting of ongoing clinical trials and completed clinical trials to public registries.
The FDA offers a number of regulatory mechanisms that provide expedited or accelerated approval procedures for selected drugs and indications which are designed to address unmet medical needs in the treatment of serious or life-threatening diseases or conditions. These include programs such as Breakthrough Therapy designations, Fast Track designations, Priority Review and Accelerated Approval, which the Corporation may need to rely upon in order to receive timely approval or to be competitive.
The Corporation may plan to seek orphan drug designation for certain indications qualified for such designation. The United States, European Union and other jurisdictions may grant orphan drug designation to drugs intended to treat a "rare disease or condition," which, in the US, is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or 200,000 or more individuals in the United States and for which there is no reasonable expectation that the cost of developing and making a drug available in the United States for this type of disease or condition will be recovered from sales of the product. In the EU, orphan drug designation can be granted if: the disease is life threatening or chronically debilitating and affects no more than 50 in 100,000 persons in the EU; without incentive it is unlikely that the drug would generate sufficient return to justify the necessary investment; and no satisfactory method of treatment for the condition exists or, if it does, the new drug will provide a significant benefit to those affected by the condition. Orphan drug designation must be requested before submitting an NDA. If a product that has an orphan drug designation subsequently receives the first regulatory approval for the indication for which it has such designation, the product is entitled to orphan exclusivity, meaning that the applicable regulatory authority may not approve any other applications to market the same drug for the same indication, except in very limited circumstances, for a period of seven years in the United States and 10 years in the EU. Orphan drug designation does not prevent competitors from developing or marketing different drugs for the same indication or the same drug for different indications. After orphan drug designation is granted, the identity of the therapeutic agent and its potential orphan use are publicly disclosed. Orphan drug designation does not convey an advantage in, or shorten the duration of, the development, review and approval process. However, this designation provides an exemption from marketing and authorization (NDA) fees.
Drugs manufactured or distributed pursuant to FDA approvals are subject to continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, reporting of adverse experiences with the product, and complying with promotion and advertising requirements. The FDA may impose a number of post-approval requirements as a condition of approval of an NDA. For example, the FDA may require post-market testing, including phase IV clinical trials, and surveillance to further assess and monitor the product's safety and effectiveness after commercialization. In addition, drug manufacturers and their subcontractors involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and certain state agencies and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with ongoing regulatory requirements, including current Good Manufacturing Practices, which impose certain procedural and documentation requirements. Failure to comply with statutory and regulatory requirements may subject a manufacturer to legal or regulatory action, such as warning letters, suspension of manufacturing, product seizures, injunctions, civil penalties or criminal prosecution. There is also a continuing, annual prescription drug product program user fee.
The FDA may withdraw approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information, requirements for post-market studies or clinical trials to assess new safety risks, or imposition of distribution or other restrictions under a risk evaluation and mitigation strategy.
In the United States, pharmaceutical manufacturers are subject to complex laws and regulations pertaining to healthcare "fraud and abuse," including, but not limited to, the Anti-Kickback Statute, the federal False Claims Act (the "FCA"), and other state and federal laws and regulations. The Anti-Kickback Statute makes it illegal for any person, including a prescription drug manufacturer (or a party acting on its behalf) to knowingly and willfully solicit, receive, offer, or pay any remuneration that is intended to induce the referral of business, including the purchase, order, or prescription of a particular drug, for which payment may be made under a federal healthcare program, such as Medicare or Medicaid.
The FCA prohibits, among other things, any person or entity from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment to or approval by the federal government or knowingly making, using, or causing to be made or used a false record or statement material to a false or fraudulent claim to the federal government. A claim includes "any request or demand" for money or property presented to the U.S. government. Violations of the FCA can result in very significant monetary penalties and treble damages. The federal government is using the FCA, and the accompanying threat of significant liability, in its investigation and prosecution of pharmaceutical companies throughout the country, for example, in connection with the promotion of products for unapproved uses and other sales and marketing practices. The government has obtained multi-million and multibillion dollar settlements under the FCA in addition to individual criminal convictions under applicable criminal statutes. In addition, the federal civil monetary penalties statute imposes penalties against any person or entity that, among other things, is determined to have presented or caused to be presented a claim to a federal health program that the person knows or should know is for an item or service that was not provided as claimed or is false or fraudulent. Given the significant size of actual and potential settlements, it is expected that the government will continue to devote substantial resources to investigating healthcare providers' and manufacturers' compliance with applicable fraud and abuse laws.
There are also an increasing number of state laws that require manufacturers to make reports to states on pricing and marketing information. In addition, a similar federal requirement Section 6002 of the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or the Affordable Care Act, commonly referred to as the "Physician Payments Sunshine Act" requires applicable manufacturers to track and report to the federal government certain payments and "transfers of value" made to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members, made in the previous calendar year. There are a number of states that have various types of additional reporting requirements.
Controlled Substances
The CSA and its implementing regulations establish a "closed system" of regulations for controlled substances. The CSA imposes registration, security, recordkeeping and reporting, storage, manufacturing, distribution, importation and other requirements under the oversight of the DEA. The DEA is responsible for regulating controlled substances, and requires those individuals or entities that manufacture, import, export, distribute, research, or dispense controlled substances to comply with the regulatory requirements in order to prevent the diversion of controlled substances to illicit channels of commerce.
The DEA categorizes controlled substances into one of five schedules - Schedule I, II, III, IV or V - with varying qualifications for listing in each schedule. Schedule I substances by definition have a high potential for abuse, have no currently accepted medical use in treatment in the United States and lack accepted safety for use under medical supervision. For any product containing a Schedule I substance, such as psilocybin, to be available for commercial marketing in the United States, such substance must be rescheduled, or the product itself must be scheduled, by the DEA to Schedule II, III, IV or V. Scheduling determinations by the DEA are dependent on FDA approval of a substance or a specific formulation of a substance.
Facilities that manufacture, distribute, import or export any controlled substance must register annually with the DEA. The DEA registration is specific to the particular location, activity(ies) and controlled substance schedule(s). For example, separate registrations are needed for import and manufacturing, and each registration will specify which schedules of controlled substances are authorized.
The DEA inspects all manufacturing facilities to review security, recordkeeping, reporting and handling prior to issuing a controlled substance registration and periodically to ensure continued compliance. The specific security requirements vary by the type of business activity and the schedule and quantity of controlled substances handled. The most stringent requirements apply to manufacturers of Schedule I and Schedule II substances. Required security measures commonly include background checks on employees and physical control of controlled substances through storage in approved vaults, safes and cages, and through use of alarm systems and surveillance cameras. Once registered, manufacturing facilities must maintain records documenting the manufacture, receipt and distribution of all controlled substances. Manufacturers must submit periodic reports to the DEA of the distribution of Schedule I and II controlled substances, Schedule III narcotic substances, and other designated substances. Registrants must also report any controlled substance thefts or significant losses, and must obtain authorization to destroy or dispose of controlled substances. Imports of Schedule I and II controlled substances for commercial purposes are generally restricted to substances not already available from a domestic supplier or where there is not adequate competition among domestic suppliers. In addition to an importer or exporter registration, importers and exporters must obtain a permit for every import or export of a Schedule I and II substance or Schedule III, IV and V narcotic, and submit import or export declarations for Schedule III, IV and V non-narcotics.
For drugs manufactured in the United States, the DEA establishes annually an aggregate quota for the amount of substances within Schedules I and II that may be manufactured or produced in the United States based on the DEA's estimate of the quantity needed to meet legitimate medical, scientific, research and industrial needs. The quotas apply equally to the manufacturing of the active pharmaceutical ingredient and production of dosage forms. The DEA may adjust aggregate production quotas a few times per year, and individual manufacturing or procurement quotas from time to time during the year, although the DEA has substantial discretion in whether or not to make such adjustments for individual companies.
Individual U.S. states also establish and maintain separate controlled substance laws and regulations, including licensing, recordkeeping, security, distribution, and dispensing requirements. A majority of state laws in the United States classify psilocybin and psilocin as Schedule I controlled substances. State authorities, including boards of pharmacy, regulate use of controlled substances in each state. Failure to maintain compliance with applicable requirements, particularly as manifested in the loss or diversion of controlled substances, can result in enforcement action that could have a material adverse effect on the Corporation's business, operations and financial condition. The DEA may seek civil penalties, refuse to renew necessary registrations, or initiate proceedings to revoke those registrations. In certain circumstances, violations could lead to criminal prosecution.
Netherlands
Regulation (EU) No 536/2014 on Clinical Trials on Medicinal Products for Human Use ( the "CTR") is applicable as of January 31, 2022, harmonizing the laws, regulations and administrative provisions of the EU Member States relating to the implementation of Good Clinical Practice in the conduct of clinical trials on medicinal products for human use. EU Member States have transformed the requirements outlined in the Clinical Trials Directive into the respective national laws. Pursuant to the CTR, as of January 31, 2023 sponsors are obliged to use the Clinical Trials Information System (CTSI) for regularity submission, authorization and supervision of clinical trials in the EU and the EEA. CTIS will thus serve as the single-entry point for submissions by sponsors and for regulatory assessment. In addition to this obligation, sponsors must transfer any ongoing (approved) trials under the CTR to CTIS by January 2025.
The IMPD is one of several regulatory documents required for conducting a clinical trial of a pharmacologically API intended for one or more EU Member States. The IMPD includes summaries of information related to the quality, manufacture and control of any Investigational Medicinal Product (including reference product and placebo) ("IMP"), and data from non-clinical and clinical studies. Guidance concerning IMPDs is based on the CTR and on the approximation of laws, regulations and administrative provisions of the Member States relating to the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use (also commonly referred to as the "Clinical Trials Directive").
The content of the IMPD may be adapted to the existing level of knowledge and the product's phase of development. When applying for a clinical trial authorization, a full IMPD is required when little or no information about an API has been previously submitted to competent authorities, when it is not possible to cross-refer to data submitted by another sponsor and/or when there is no authorization for sale in the EU. However, a simplified IMPD may be submitted if information has been assessed previously as part of a Marketing Authorization or a clinical trial to that competent authority. Although the format is not obligatory, the components of an IMPD are largely equivalent to clinical trial applications in Canada and the United States. The IMPD need not be a large document as the amount of information to be contained in the dossier is dependent on various factors such as product type, indication, development phase etc.
The assessment of an IMPD is focused on patient safety and any risks associated with the IMP. Whenever any potential new risks are identified the IMPD must be amended to reflect the changes. Certain amendments are considered substantial in which case the competent authority must be informed of the substantial amendment. This may be the case for changes in IMP impurities, microbial contamination, viral safety, transmissible spongiform encephalopathies (e.g. mad cow disease) and in some particular cases to stability when toxic degradation products may be generated.
With the completion of the Asset Acquisition, the Corporation has an ongoing phase I study to obtain preliminary evidence of the safety and efficacy of infused DMT. Prior to the Asset Acquisition, an investigator's brochure (including prior safety, preclinical and clinical data), and an IMPD document that includes CMC information and a clinical study protocol and supporting information had been prepared. Approval by the Dutch ethics committee of the Phase 1 Study, planned to be conducted by CHDR will be based on the vast amount of published human and animal studies of DMT. Prior to the Asset Acquisition, preclinical data was not provided as part of the application package; however, limited additional in vivo and in vitro data to support the rationale for human dosing and safety had been included. CHDR and its partner GMP-licensed pharmacy that will be involved in the Phase 1 Study, the Leiden University Medical Center, have all the required approvals to possess and handle DMT for the Phase 1 Study.
Failure of the Corporation to receive the necessary regulatory approvals required to conduct the Phase 1 Study would have an adverse impact on its business plans and financial condition for a number of reasons including, without limitation: (i) it would cause delays in the Corporation's research and development plans; (ii) it may require the Corporation to expend additional financial and human resources on revising its application package or creating a new one; or (iii) it may require the Corporation to approach an entirely different regulatory authority in a new jurisdiction, in which case the Corporation would have to expend a substantial amount of capital and other resources on engaging the appropriate research and development partners and creating an application package that complies with the regulations of that new jurisdiction. Additionally, the Corporation would be required to spend capital on transferring the DMT materials to the new jurisdiction. All of the foregoing would likely have a negative impact on the Corporation's business and financial condition.
Pharmaceutical Products
In accordance with the Dutch Medicines Act (Geneesmiddelenwet), "medicinal products" are defined as: a substance or a combination of substances that is intended to be administered or used for, or is presented in any way as being suitable for, use: (i) the cure or prevention of any disease, defect, wound or pain in human beings, (ii) the making of a medical diagnosis in human beings, or (iii) restoring, improving or otherwise modifying physiological functions in humans by exerting a pharmacological, immunological or metabolic effect.
If a product constitutes a medicinal product, a marketing authorization for the product is required before the product may be placed on the market in the Netherlands. In the EU, marketing authorizations may be obtained through the Centralized procedure, the Decentralized procedure and/or the national procedure. The Centralized procedure is compulsory for medicines intended to treat i.e. cancer, AIDS, neurodegenerative diseases and diabetes and optional (only) for medicines comprising of new active substances not previously approved for the EEA. When applying for a marketing authorization through the Centralized procedure, applications are submitted with the European Medicines Agency (the "EMA"). Where the Centralized procedure is not available but a medicinal product is intended for several EU/EEA Member States, an application for a marketing authorization may be submitted with the competent authority of a single EU/EEA Member State in accordance with the Decentralized procedure. When the assessment of the application results in a decision to grant the marketing authorization, this decision will be mutually recognized by the competent authorities of the other Member States for which the marketing authorization is applied. Finally, should a medicinal product be intended for the Netherlands only, then the national procedure may be followed as well by submitting an application with the Dutch Medicines Evaluation Board. It may be remarked that the national procedure is unavailable in case the Centralized procedure is compulsory or in case an applicant has already submitted an application for and/or obtained a marketing authorization in another Member State. In that case, applications must follow the mutual recognition procedure instead.
Companies that manufacture or trade in medicinal products and/or active pharmaceutical ingredients in the Netherlands require a manufacturing authorization or a wholesale distribution authorization. A manufacturing authorization is required for the preparation, trading in, import and export of medicinal products and/or active substances. Here, 'preparation' means the total or partial manufacture of medicinal products and/or active substances or the packaging or labelling thereof. 'Importing' means the import of medicinal products or active substances from a country outside the EEA into the Dutch territory, while 'exporting' means the export of medicinal products or active substances from the Dutch territory to a country outside the EEA. A wholesale distribution authorization is required for one or more activities within the wholesale business, such as procuring, holding, supplying, delivering or exporting medicinal products or active substances which are prepared or imported by a third party. It may be noted that holders of wholesale distribution authorization, other than holders of marketing authorizations, are not authorized to import medicinal products from countries outside the EEA.
Only a natural or legal person established in the Netherlands may obtain either a Dutch marketing authorization or a wholesale distribution authorization. These authorizations concern national permits, meaning that these authorizations are not automatically valid in other EU Member States. Furthermore, in the Netherlands applicants of marketing authorizations and wholesale distributions authorizations must be registered with Farmatec and comply with GDP norms.
Market Authorization Regulatory Process
Under the Centralized procedure, pharmaceutical companies submit a single marketing authorization application to the EMA, which provides the basis of a legally binding recommendation that will be provided by the EMA to the European Commission, the authorizing body for all centrally authorized products. This allows the marketing authorization holder to market the medicine and make it available to patients and healthcare professionals throughout the EU on the basis of a single marketing authorization. EMA's Committee for Medicinal products for Human Use or Committee for Medicinal Products for Veterinary Use carry out a scientific assessment of the application and give a recommendation on whether the medicine should be marketed or not, under any particular dosing regime. Although, under EU law, the EMA has no authority to permit marketing in the different EU countries, the European Commission is the authorizing body for all centrally authorized products, who takes a legally binding decision based on EMA's recommendation. Once granted by the European Commission, the centralized marketing authorization is valid in all EU Member States as well as in the European Economic Area countries Iceland, Liechtenstein and Norway. European Commission decisions are published in the Community Register of medicinal products for human use. Once a medicine has been authorized for use in the EU, the EMA and the EU Member States constantly monitor its safety and take action if new information indicates that the medicine is no longer as safe and effective as previously thought. The safety monitoring of medicines involves a number of routine activities ranging from: assessing the way risks associated with a medicine will be managed and monitored once it is authorized; continuously monitoring suspected side effects reported by patients and healthcare professionals, identified in new clinical studies or reported in scientific publications; regularly assessing reports submitted by the Corporation holding the marketing authorization on the benefit-risk balance of a medicine in real life; and assessing the design and results of post-authorization safety studies which were required at the time of authorization. The EMA can also carry out a review of a medicine or a class of medicines upon request of a Member State or the European Commission. These are called EU referral procedures; they are usually triggered by concerns in relation to a medicine's safety, the effectiveness of risk minimization measures or the benefit-risk balance of the medicine. The EMA has a dedicated committee responsible for assessing and monitoring the safety of medicines, the Pharmacovigilance Risk Assessment Committee. This ensures that EMA and the EU Member States can move very quickly once an issue is detected and take any necessary action, such as amending the information available to patients and healthcare professionals, restricting use or suspending a medicine, in a timely manner in order to protect patients.
Besides the Centralized procedure, pharmaceutical companies may also submit marketing authorization applications through the Decentralized procedure with the competent authority of a Member State. As the Centralized procedure is compulsory for medicines intended to treat specified diseases i.e. cancer, AIDS, neurodegenerative diseases and diabetes and only optional for medicines comprising of new active substances not previously approved for the EU/ EEA, in all other circumstances the Decentralized procedure should be used instead if a marketing authorization is to be obtained for several EU/EEA Member States. When following the Decentralized procedure, the applicant requests one country to be the Reference Member State ("RMS") in the procedure. After having shared draft assessment reports to which both the applicant and the competent authorities of other Member States may respond, the to be granted marketing authorization will eventually go through the Mutual recognition procedure. In the Mutual recognition procedure other Member States generally adopt the RMS's assessment, unless there are important objections on the grounds of a potentially serious risk to public health. In such situations, further discussions will also be held in the Co-ordination group for Mutual recognition and Decentralised procedures ("CMDh"). When all Member States involved decide on a positive opinion on products in the CMDh, Dutch translations of the summary of product characteristics, package leaflet, labelling texts and mock-ups are submitted and a national marketing authorization is issued.
Patent Cooperation Treaty
The Patent Cooperation Treaty (the "PCT") facilitates filing for patent recognition in multiple jurisdictions simultaneously using a single uniform patent application. 157 countries, including Canada and the United States have ratified the PCT.
Ultimately, patents are still granted in each country individually. As such, the PCT procedure consists of two phases: filing of an international application, and national evaluation under the patent laws in force in each country where a patent is sought.
Within 12 months of filing a provisional patent application at the United States Patent and Trademark Office, the Corporation may elect to file a regular utility patent application in the United States in tandem with filing a PCT application with the World Intellectual Property Office, in each case claiming priority to the provisional patent application. Within 30 months of the provisional filing date, deadlines begin for a PCT application to enter the national phase in desired jurisdictions globally, such as Canada (30 months) and Europe (31 months), in each case claiming priority to the provisional patent application.
While the Corporation is focused on programs using psychedelic-inspired compounds, the Corporation does not have any direct or indirect involvement with the illegal selling, production or distribution of any substances in the jurisdictions in which it operates. The Corporation is exploring drug development within approved laboratory clinical trial settings conducted within approved regulatory frameworks. Though highly speculative, should any prescription drug product be developed by the Corporation (which, if it does occur, would not be for several years), such drug product will not be commercialized prior to receipt of applicable regulatory approval, which will only be granted if clinical evidence of safety and efficacy for the intended use(s) is successfully developed. The Corporation may also employ non-prescription drugs, where appropriate.
COMPLIANCE PROGRAM
The Corporation oversees and monitors compliance with applicable laws in each jurisdiction in which it operates. In addition to the Corporation's senior executives and the employees responsible for overseeing compliance, the Corporation has local counsel engaged in every jurisdiction in which it operates and has received legal opinions or advice in each of these jurisdictions regarding (a) compliance with applicable regulatory frameworks, and (b) potential exposure to, and implications arising from, applicable laws in jurisdictions in which the Corporation has operations or intends to operate.
The Corporation works with third parties who require regulatory licensing to handle scheduled drugs. The Corporation continuously updates its compliance and channel programs to maintain regulatory standards set for drug development. The Corporation also works with clinical research organizations who maintain batch records and data storage for the Corporation's clinical programs.
Additionally, the Corporation has established a Medical & Clinical Advisory Team, a Research, Clinical and Regulatory Team and a Government Relations and Communications Team with cross-functional expertise in business, neuroscience, pharmaceuticals, mental health and psychedelics to advise management.
In conjunction with the Corporation's human resources and operations departments, the Corporation oversees and implements training on the Corporation's protocols. The Corporation will continue to work closely with external counsel and other compliance experts, and is evaluating the engagement of one or more independent third party providers to further develop, enhance and improve its compliance and risk management and mitigation processes and procedures in furtherance of continued compliance with the laws of the jurisdictions in which the Corporation operates.
The programs currently in place include monitoring by executives of the Corporation to ensure that operations conform to and comply with required laws, regulations and operating procedures. The Corporation is currently in compliance with the laws and regulations in all jurisdictions and the related licencing framework applicable to its business activities.
The Corporation and, to its knowledge, each of its third-party researchers, suppliers and manufacturers have not received any non-compliance, citations or notices of violation which may have an impact on the Corporation's licences, business activities or operations.
The Corporation conducts due diligence on third-party researchers, medical professionals, clinics, cultivators, processors and others as applicable, with whom it engages. Such due diligence includes but is not limited to the review of necessary licenses and the regulatory framework enacted in the jurisdiction of operation. Further, the Corporation generally obtains, under its contractual arrangements, representations and warranties from such third parties pertaining to compliance with applicable licensing requirements and the regulatory framework enacted in the jurisdiction of operation.
CONSOLIDATED CAPITALIZATION
There have been no material changes to the share and loan capitalization of the Corporation since the date of the Interim Financial Statements.
The applicable Prospectus Supplement will describe any material changes, and the effect of such material changes, on the share and loan capitalization of the Corporation that will result from the issuance of Securities pursuant to each Prospectus Supplement.
USE OF PROCEEDS
The net proceeds from any Offering of Securities and the proposed use of those proceeds will be set forth in the applicable Prospectus Supplement relating to that Offering of Securities. Notwithstanding, the Corporation's management has broad discretion in the application of proceeds of an Offering of Securities. On the basis of results obtained or for other sound business reasons, the Corporation may re-allocate funds as required. Accordingly, the Corporation's actual use of proceeds may vary significantly from any proposed use of proceeds disclosed in any applicable Prospectus Supplement. See "Risk Factors - Risks Related to an Offering - Discretion over the Use of Proceeds".
Sources and Uses of Capital
As at August 8, 2023, the Corporation had cash resources, being cash and an HST receivable, of approximately $21,082,000. As at August 8, 2023, the Corporation anticipates that it will require approximately $21,082,000 to continue operations over the next 12 months, including funding for the achievement of the next significant milestone under the Corporation’s Deuterated Psilocybin Analog Program (CYB003) and the Deuterated Dimethyltryptamine Program (CYB004).
The Corporation may be required to reduce its general and administrative expenses if it does not have sufficient capital to meet its below stated objectives, or it may allocate its financial resources differently than listed below. The expected reduction in future expenditures, absent any capital injections, compared to the Corporation’s historic expenditures would be achieved as follows: (i) a reduction in marketing activities; (ii) a reduction in workforce and/or compensation; (iii) a reduction in public relations and investor relations activities; and (iv) a reduction of planned expenses under the Deuterated Dimethyltryptamine Program and Phenethylamine Derivatives Program, which would also result in a delay of anticipated timing for completion of the next significant milestones under each of these programs.
| Program(1) |
Anticipated
12 Month Use
of Funds(2)(3)
($) |
|
Anticipated
Timeline for
Completio(4) |
| Deuterated Psilocybin Analog Program |
|
|
|
| Provide topline data readout from the Phase 1/2a study |
3,880,627 |
|
Q3/Q4 2023 |
| Complete FDA submission of Phase 1/2a data |
2,500,200 |
|
Q4 2023 |
| Deuterated Dimethyltryptamine Program |
-
|
|
-
|
| Provide topline data from Phase 1 CYB004-E trial |
1,269,000 |
|
Q3/Q4 2023 |
| Complete FDA IND submission |
1,372,950 |
|
Q1 2024 |
| Phenethylamine Derivatives Program |
-
|
|
-
|
| Technology Programs |
-
|
|
-
|
| Working Capital, and General Corporate Purposes(5) |
12,059,223
|
|
N/A
|
| Total |
21,082,000
|
|
|
Notes:
(1) Please see the Corporation’s Interim MD&A for a description of the Corporation’s programs. All milestones are subject to receipt of all necessary approvals, including the academic and scientific organizations with which the Corporation is working.
(2) Certain amounts have been converted from USD to CAD at an exchange rate of 1.35:1.
(3) Represents proceeds from private placements and offerings previously disclosed, as well proceeds from the ATM Program and May 2023 Prospectus.
(4) Anticipated timeline for completion is based on the calendar year. Anticipated spending and timelines regarding drug development are based on reasonable assumptions informed by current knowledge and information available to the Corporation.
(5) Includes personnel costs, professional services, overhead expenses and general expenses to be incurred by the Corporation in the normal course of business. In addition, the Corporation intends to use a portion of these proceeds to continue funding both its Deuterated Psilocybin Analog Program and Deuterated Dimethyltryptamine Program. The allocation between and specific milestones within these programs have not yet been determined.
For detailed information in respect of the Corporation’s business objectives and milestones, and the application of proceeds from prior offerings by the Corporation, prospective purchasers of Securities should carefully consider the information described in the Annual Information Form and Interim MD&A of the Corporation, and the documents incorporated by reference herein, including the applicable Prospectus Supplement.
As at August 8, 2023, the Corporation’s current financial resources, absent any capital injections, will be sufficient to meet the Corporation’s short-term liquidity requirements and to fund its operations for the coming 12 months. The Corporation makes this statement based on its cash resources, being cash and an HST receivable, as of August 8, 2023 in the amount of approximately $21,082,000.
The anticipated spend and cash burn do not account for funds that the Corporation had previously disclosed as being required to progress its Phenethylamine Derivatives Program listed in the table above. As disclosed in its Interim MD&A, the Corporation anticipated spending approximately $900,000 under its Phenethylamine Derivatives Program by September 30, 2023 to complete preclinical development of a phenethylamine drug candidate, of which approximately $42,000 was spent during the three months ended June 30, 2023, and approximately $782,000 was spent during the financial year ended March 31, 2023, resulting in an approximate remaining spend as of June 30, 2023 of $76,000 by September 30, 2023.10 If the Corporation does not raise additional funds in the coming 12 months, it anticipates that it will need to reduce the amounts spent on the Phenethylamine Derivatives Program programs and adjust the corresponding timelines disclosed in its Interim MD&A.
_____________________________________________
10 Reflects actual spend during the financial year ended March 31, 2023, the three months ended June 30, 2023, and expected spend during the period from July 1, 2023 until the achievement of preclinical development of a phenethylamine drug candidate by September 30, 2023. The Company had previously estimated that its spending to complete preclinical development of a phenethylamine drug candidate would be $1,283,000. Anticipated timing and spending regarding drug development is based on reasonable assumptions informed by current knowledge and information available to the Corporation. Such statements are informed by, among other things, regulatory guidelines for developing a drug with safety studies, proof of concept studies, and pivotal studies for new drug application submission and approval, and assumes the success of implementation and results of such studies on timelines indicated as possible by such guidelines, other industry examples, and the Corporation’s development efforts to date.
The Corporation's expectation is based on certain assumptions and risks as set forth in the section "Cautionary Note Regarding Forward Looking Information" and "Risk Factors" in this Prospectus.
The expected uses of capital represents the Corporation's current intentions based upon its present plans and business condition, which could change in the future as its plans and business conditions evolve. The amounts and timing of the actual use of available capital will depend on multiple factors and there may be circumstances where, for sound business reasons, a reallocation of capital, or termination of a program objective, may be necessary in order for the Corporation to achieve its program objectives. The Corporation may also require additional funds in order to fulfill its expenditure requirements to meet existing and any new business objectives, and the Corporation expects to either issue additional securities or incur debt to do so. The material factors or assumptions used to develop the estimated amounts for the 12 months period disclosed above are included in the "Cautionary Note Regarding Forward-Looking Information" section above. The actual amount that the Corporation spends in connection with each of the identified uses and programs will depend on a number of factors, including those listed under "Risk Factors" in, or incorporated by reference in, this Prospectus.
Negative Cash Flow From Operations
Since inception, the Corporation has financed its operations primarily from the issuance of equity and interest income on funds available for investment. To date, the Corporation has raised approximately C$156,500,000 in gross proceeds through private placement and prospectus offerings. The Corporation has experienced operating losses and cash outflows from operations since incorporation and will require ongoing financing to continue its research and development activates. As the Corporation has not yet achieved profitability, there are uncertainties regarding its ability to continue as a going concern. The Corporation has not earned any revenue or reached successful commercialization of any products. The Corporation's success is dependent upon the ability to finance its cash requirements to continue its activities. There is no assurance that additional capital or other types of financing will be available if needed or that these financings will be on terms at least as favourable to the Corporation as those previously obtained, or at all. To the extent that the Corporation has negative operating cash flows in future periods, it may need to deploy a portion of its existing working capital to fund such negative cash flows. The Corporation will be required to raise additional funds through the issuance of additional equity securities, through loan financing, or other means, such as through partnerships with other companies and research and development reimbursements. There is no assurance that additional capital or other types of financing will be available if needed or that these financings will be on terms at least as favourable to the Corporation as those previously obtained. See "Risk Factors - Risks Related to an Offering - Negative Operating Cash Flow and Going Concern".
DIVIDEND POLICY
The Corporation has never declared nor paid dividends on the Common Shares. Currently, the Corporation intends to retain its future earnings, if any, to fund the development and growth of its business, and the Corporation does not anticipate declaring or paying any dividends on the Common Shares in the near future, although the Corporation reserves the right to pay dividends if and when it is determined to be advisable by the board of directors of the Corporation. As a result, shareholders will have to rely on capital appreciation, if any, to earn a return on investment in the Common Shares in the foreseeable future. See "Risk Factors - Risks Related to an Offering - Speculative Nature of Investment Risk".
DESCRIPTION OF SECURITIES BEING DISTRIBUTED
The following is a brief summary of certain general terms and provisions of the Securities that may be offered pursuant to this Prospectus. This summary does not purport to be complete. The particular terms and provisions of the Securities as may be offered pursuant to this Prospectus will be set forth in the applicable Prospectus Supplement pertaining to such Offering of Securities, and the extent to which the general terms and provisions described below may apply to such Securities will be described in the applicable Prospectus Supplement.
Common Shares
The Corporation is authorized to issue an unlimited number of Common Shares and an unlimited number of preferred shares. As at August 16, 2023, the Corporation had 234,683,234 Common Shares and nil preferred shares issued and outstanding.
Each Common Share entitles the holder thereof to one vote at meetings of shareholders of the Corporation other than meetings of the holders of another class of shares. Each holder of Common Shares is also entitled to receive dividends if, as and when declared by the board of directors of the Corporation. Holders of Common Shares are entitled to participate in any distribution of the Corporation's net assets upon liquidation, dissolution or winding-up on an equal basis per share. There are no pre-emptive, redemption, retraction, purchase or conversion rights attaching to the Common Shares.
Common Shares may be sold separately or together with certain other Securities under this Prospectus. Common Shares may also be issuable on conversion, exchange, exercise or maturity of certain other Securities qualified for issuance under this Prospectus.
Warrants
Warrants may be offered separately or together with other Securities, as the case may be. Each series of Warrants may be issued under a separate warrant indenture or warrant agency agreement to be entered into between the Corporation and one or more banks or trust companies acting as Warrant agent or may be issued as stand-alone contracts. The applicable Prospectus Supplement will include details of the Warrant agreements, if any, governing the Warrants being offered. The Warrant agent, if any, will be expected to act solely as the agent of the Corporation and will not assume a relationship of agency with any holders of Warrant certificates or beneficial owners of Warrants. The following sets forth certain general terms and provisions of the Warrants that may be offered under this Prospectus. The specific terms of the Warrants, and the extent to which the general terms described in this section apply to those Warrants, will be set forth in the applicable Prospectus Supplement.
A copy of any warrant indenture or any warrant agency agreement relating to an offering of Warrants will be filed by the Corporation with the relevant securities regulatory authorities in Canada after it has been entered into by the Corporation.
Each applicable Prospectus Supplement will set forth the terms and other information with respect to the Warrants being offered thereby, which may include, without limitation, the following (where applicable):
-
the designation of the Warrants;
-
the aggregate number of Warrants offered and the offering price;
-
the designation, number and terms of the other Securities purchasable upon exercise of the Warrants, and procedures that will result in the adjustment of those numbers;
-
the exercise price of the Warrants;
-
the dates or periods during which the Warrants are exercisable;
-
the designation and terms of any securities with which the Warrants are issued;
-
if the Warrants are issued as a unit with another Security, the date on and after which the Warrants and the other Security will be separately transferable;
-
any minimum or maximum amount of Warrants that may be exercised at any one time;
-
whether such Warrants will be listed on any securities exchange;
-
any terms, procedures and limitations relating to the transferability, exchange or exercise of the Warrants;
-
certain material Canadian tax consequences of owning the Warrants; and
-
any other material terms and conditions of the Warrants.
Units
The Corporation may issue Units comprised of one or more of the other Securities described herein in any combination. Each Unit may be issued so that the holder of the Unit is also the holder of each Security included in the Unit; thus, the holder of a Unit may have the rights and obligations of a holder of each included Security. Any Unit agreement under which a Unit may be issued may provide that the Securities included in the Unit may not be held or transferred separately at any time or at any time before a specified date.
Each applicable Prospectus Supplement will set forth the terms and other information with respect to the Units being offered thereby, which may include, without limitation, the following (where applicable):
-
the designation, number and terms of the Units and of the Securities comprising the Units, including whether and under what circumstances those Securities may be held or transferred separately;
-
any provisions for the issuance, payment, settlement, transfer or exchange of the Units or of the Securities comprising the Units;
-
certain material Canadian tax consequences of owning the Securities comprising the Units; and
-
any other material terms and conditions of the Units.
Debt Securities
The Debt Securities may be issued in one or more series under an indenture (the "Indenture") to be entered into between the Corporation and one or more trustees (the "Trustee") that may be named in a Prospectus Supplement for a series of Debt Securities. To the extent applicable, the Indenture will be subject to and governed by the United States Trust Indenture Act of 1939, as amended. A copy of the form of the Indenture to be entered into has been or will be filed with the SEC as an exhibit to the Registration Statement of which this Prospectus forms a part and will be filed with the securities commissions or similar authorities in Canada when it is entered into. The Corporation may issue Debt Securities, separately or together, with Common Shares, Warrants, Units or Subscription Receipts or any combination thereof, as the case may be.
The description of certain provisions of the Indenture in this section do not purport to be complete and are subject to, and are qualified in their entirety by reference to, the provisions of the Indenture. The following sets forth certain general terms and provisions of the Debt Securities. The particular terms and provisions of a series of Debt Securities offered pursuant to this Prospectus will be set forth in the applicable Prospectus Supplement, and the extent to which the general terms and provisions described below may apply to such Debt Securities will be described in the applicable Prospectus Supplement. This description may include, but may not be limited to, any of the following, if applicable:
-
the title of the Debt Securities;
-
any limit on the aggregate principal amount of the Debt Securities;
-
the date or dates, if any, on which the Debt Securities will mature and the portion (if less than all of the principal amount) of the Debt Securities to be payable upon declaration of acceleration of maturity;
-
the rate or rates (whether fixed or variable) at which the Debt Securities will bear interest, if any, the date or dates from which any such interest will accrue and on which any such interest will be payable and the record dates for any interest payable on the Debt Securities;
-
the terms and conditions under which the Corporation may be obligated to redeem, repay or purchase the Debt Securities pursuant to any sinking fund or analogous provisions or otherwise;
-
the terms and conditions upon which the Corporation may redeem the Debt Securities, in whole or in part, at its option;
-
the covenants applicable to the Debt Securities;
-
the terms and conditions for any conversion or exchange of the Debt Securities for any other securities;
-
the extent and manner, if any, to which payment on or in respect of the Debt Securities of the series will be senior or will be subordinated to the prior payment of other liabilities and obligations of the Corporation;
-
whether the Debt Securities will be secured or unsecured;
-
whether the Debt Securities will be issuable in the form of global securities ("Global Securities"), and, if so, the identity of the depositary for such Global Securities;
-
the denominations in which Debt Securities will be issuable, if other than denominations of US$1,000 or integral multiples of US$1,000;
-
each office or agency where payments on the Debt Securities will be made and each office or agency where the Debt Securities may be presented for registration of transfer or exchange;
-
if other than United States dollars, the currency in which the Debt Securities are denominated or the currency in which we will make payments on the Debt Securities;
-
material Canadian federal income tax consequences and United States federal income tax consequences of owning the Debt Securities; and
-
any other terms, conditions, rights or preferences of the Debt Securities which apply solely to the Debt Securities.
If the Corporation denominates the purchase price of any of the Debt Securities in a currency or currencies other than United States dollars or a non-United States dollar unit or units, or if the principal of and any premium and interest on any Debt Securities is payable in a currency or currencies other than United States dollars or a non-United States dollar unit or units, the Corporation will provide investors with information on the restrictions, elections, general tax considerations, specific terms and other information with respect to that issue of Debt Securities and such non-United States dollar currency or currencies or non-United States dollar unit or units in the applicable Prospectus Supplement.
This Prospectus does not qualify for issuance Debt Securities in respect of which the payment of principal and/or interest may be determined, in whole or in part, by reference to one or more underlying interests including, for example, an equity or debt security, a statistical measure of economic or financial performance including, but not limited to, any currency, consumer price or mortgage index, or the price or value of one or more commodities, indices or other items, or any other item or formula, or any combination or basket of the foregoing items. For greater certainty, this Prospectus may qualify for issuance Debt Securities in respect of which the payment of principal and/or interest may be determined, in whole or in part, by reference to published rates of a central banking authority or one or more financial institutions, such as a prime rate or bankers’ acceptance rate, or to recognized market benchmark interest rates such as CORRA, SOFR, EURIBOR or an alternative United States federal funds rate.
Each series of Debt Securities may be issued at various times with different maturity dates, may bear interest at different rates and may otherwise vary.
The terms on which a series of Debt Securities may be convertible into or exchangeable for Common Shares or other securities of the Corporation will be described in the applicable Prospectus Supplement. These terms may include provisions as to whether conversion or exchange is mandatory, at the option of the holder or at the option of the Corporation, and may include provisions pursuant to which the number of Common Shares or other securities to be received by the holders of such series of Debt Securities would be subject to adjustment.
To the extent any Debt Securities are convertible into Common Shares or other securities of the Corporation, prior to such conversion the holders of such Debt Securities will not have any of the rights of holders of the securities into which the Debt Securities are convertible, including the right to receive payments of dividends or the right to vote such underlying securities.
The Corporation may, from time to time, issue Debt Securities and incur additional indebtedness other than through the issue of Debt Securities pursuant to this Prospectus
Subscription Receipts
Subscription Receipts may be offered separately or together with other Securities, as the case may be. The Subscription Receipts may be issued under a subscription receipt agreement.
The applicable Prospectus Supplement will include details of any subscription receipt agreement covering the Subscription Receipts being offered. A copy of any subscription receipt agreement relating to an offering of Subscription Receipts will be filed by the Corporation with the relevant securities regulatory authorities in Canada after the Corporation has entered into it. The specific terms of the Subscription Receipts, and the extent to which the general terms described in this section apply to those Subscription Receipts, will be set forth in the applicable Prospectus Supplement. This description may include, without limitation, the following (where applicable):
-
the number of Subscription Receipts;
-
the price at which the Subscription Receipts will be offered;
-
the terms, conditions and procedures for the conversion of the Subscription Receipts into other Securities;
-
the designation, number and terms of the other Securities that may be exchanged upon conversion of each Subscription Receipt;
-
the designation, number and terms of other Securities with which the Subscription Receipts will be offered, if any, and the number of Subscription Receipts that will be offered with each Security;
-
terms applicable to the gross or net proceeds from the sale of the Subscription Receipts plus any interest earned thereon;
-
certain material Canadian tax consequences of owning the Subscription Receipts; and
-
any other material terms and conditions of the Subscription Receipts.
PLAN OF DISTRIBUTION
General
The Corporation may from time to time during the 25-month period that this Prospectus, including any amendments and supplements hereto, remains valid, offer for sale and sell up to an aggregate of $160,000,000 in Securities hereunder.
The Securities may be sold by the Corporation (i) directly pursuant to applicable statutory exemptions, (ii) to or through underwriters or dealers, or (iii) through designated agents. The Prospectus Supplement relating to a particular Offering of Securities will identify any underwriter, dealer or agent engaged in connection with the offering and sale of such Securities, and will set forth the terms of the offering of such Securities, including, to the extent applicable, any fees, discounts or any other compensation payable to underwriters, dealers or agents in connection with the offering, the method of distribution of the Securities, the purchase price of the Securities (or the manner of determination thereof if offered on a non-fixed price basis), the net proceeds to the Corporation and any other material terms of the plan of distribution (including sales in transactions that are deemed to be "at-the-market distributions" as defined in NI 44-102). Any initial offering price and discounts, concessions or commissions allowed or re-allowed or paid to underwriters, dealers or agents may be changed from time to time. Only underwriters named in the Prospectus Supplement are deemed to be underwriters in connection with the Corporation's Securities offered by that Prospectus Supplement.
The Securities may be sold from time to time in one or more transactions at a fixed price or prices or at non-fixed prices. If offered on a non-fixed price basis, the Securities may be offered at market prices prevailing at the time of sale, at prices determined by reference to the prevailing price of a specified security in a specified market or at prices to be negotiated with purchasers including sales in transactions that are deemed to be "at-the-market" distributions, including sales made directly on the NEO or other existing trading markets for the Securities, in which case the compensation payable to an underwriter, dealer or agent in connection with any such sale will be decreased by the amount, if any, by which the aggregate price paid for the Securities by the purchasers is less than the gross proceeds paid by the underwriter, dealer or agent to the Corporation. The price at which the Securities will be offered and sold may vary from purchaser to purchaser and during the period of distribution.
Sales of Securities under an "at-the-market distribution", if any, will be made pursuant to an accompanying Prospectus Supplement. Sales of Securities under any "at-the-market" program will be made in transactions that are "at-the-market distributions" as defined in NI 44-102. The volume and timing of any "at-the-market distributions" will be determined at the Corporation's sole discretion.
No underwriter or dealer involved in an "at-the-market distribution" under this Prospectus, no affiliate of such an underwriter or dealer and no person or company acting jointly or in concert with such an underwriter or dealer will over-allot securities in connection with such distribution or effect any other transactions that are intended to stabilize or maintain the market price of the offered Securities or securities of the same class as the Securities distributed under the "at-the-market distribution", including selling an aggregate number or principal amount of Securities that would result in the underwriter creating an over-allocation position in the Securities.
In connection with the sale of the Securities, underwriters, dealers or agents may receive compensation from the Corporation including in the form of underwriters', dealers' or agents' fees, commissions or concessions.
Underwriters, dealers and agents that participate in the distribution of the Securities may be deemed to be underwriters for the purposes of applicable Canadian securities legislation and any such compensation that they receive from the Corporation and any profit that they make on the resale of the Securities, may be deemed to be underwriting commissions.
Underwriters, dealers or agents who participate in the distribution of the Securities may be entitled, under agreements to be entered into with the Corporation to indemnification by the Corporation against certain liabilities, including liabilities under Canadian securities legislation, or to contribution with respect to payments, which such underwriters, dealers or agents may be required to make in respect thereof. Such underwriters, dealers and agents may be customers of, engage in transactions with, or perform services for, the Corporation in the ordinary course of business.
In connection with any Offering of Securities, subject to applicable laws and other than an "at-the-market distribution", the underwriters, dealers or agents, as the case may be, may over-allot or effect transactions which stabilize, maintain or otherwise affect the market price of the offered Securities at a level other than those which otherwise might prevail on the open market. Such transactions may be commenced, interrupted or discontinued at any time.
Unless specified in the applicable Prospectus Supplement, there is no market through which the Subscription Receipts, Warrants, Units and Debt Securities may be sold and purchasers may not be able to resell the Subscription Receipts, Warrants, Units and Debt Securities purchased under this Prospectus and the Prospectus Supplement. This may affect the pricing of the Subscription Receipts, Warrants, Units and Debt Securities in the secondary market, the transparency and availability of trading prices, the liquidity of the Subscription Receipts, Warrants, Units and Debt Securities and the extent of issuer regulation. See "Risk Factors".
RISK FACTORS
An investment in the Securities involves a high degree of risk and must be considered speculative due to the nature of the Corporation’s business and present stage of development. Before making an investment decision, prospective purchasers of Securities should carefully consider the information described in this Prospectus and the documents incorporated by reference herein, including the applicable Prospectus Supplement. There are certain risks inherent in an investment in the Securities, including the factors described below and under the heading “Risk Factors” in the Annual Information Form and under the heading “Risks and Uncertainties” in the Interim MD&A, and any other risk factors described herein or in a document incorporated by reference herein, which investors should carefully consider before investing. Additional risk factors relating to a specific Offering of Securities will be described in the applicable Prospectus Supplement. Some of the factors described herein, in the documents incorporated by reference herein, and/or the applicable Prospectus Supplement are interrelated and, consequently, investors should treat such risk factors as a whole. If any of the risk factors described herein, in the Annual Information Form, in another document incorporated by reference herein or in the applicable Prospectus Supplement occur, it could have a material adverse effect on the business, financial condition and results of operations of the Corporation. Additional risks and uncertainties of which the Corporation currently is unaware or that are unknown or that it currently deems to be immaterial could have a material adverse effect on the Corporation’s business, financial condition and results of operation. The Corporation cannot assure purchasers that it will successfully address any or all of these risks. There is no assurance that any risk management steps taken will avoid future loss due to the occurrence of the risks described herein, in the Annual Information Form, in the other documents incorporated by reference herein or in the applicable Prospectus Supplement or other unforeseen risks.
Risks Related to the Business of the Corporation
Ongoing Impact of the Novel Coronavirus "COVID-19"
The outbreak of COVID-19 and its eventual declaration as a pandemic by the World Health Organization ("WHO") on March 11, 2020, resulted in governments worldwide enacting emergency measures to combat the spread of the virus. These measures, which include the implementation of travel bans, self-imposed quarantine periods and social distancing, have caused material disruption to businesses globally resulting in an economic slowdown. Global equity markets have experienced significant volatility and weakness. Governments and central banks have reacted with significant monetary and fiscal interventions designed to stabilize economic conditions. The duration and impact of the COVID-19 outbreak is still unknown at this time, as is the efficacy of the government and central bank interventions. It is not possible to reliably estimate the length and severity of these developments and the impact on the financial results and condition of the Corporation in future periods. The Corporation is not currently aware of any changes in laws, regulations or guidelines, including tax and accounting requirements, arising from COVID-19 which would be reasonably anticipated to materially affect the Corporation's business.
Regulatory Risks and Uncertainties
In Canada, certain psychedelic drugs, including psilocybin, are classified as Schedule III drugs under the CDSA and as such, medical and recreational use is illegal under Canadian federal laws. In the United States, certain psychedelic drugs, including psilocybin, psilocin, DMT, and 5-Methoxy-DMT, are classified as Schedule I drugs under the CSA and the Controlled Substances Import and Export Act and as such, medical and recreational use is illegal under the U.S. federal laws. Anyone wishing to conduct research on substances listed in Schedule I under the CSA must register with the DEA and obtain DEA approval of the research proposal. The EU member states currently classify DMT as a Schedule I substance under the UN 71 and, as such, a licence is required to produce, dispense, import or export any Schedule I substances, but the specific requirements vary from country to country. Currently in the Netherlands, DMT is classified as a List 1 Drug under the Dutch Opium Act and, as such, subject to express authorization being obtained, the production, trade and possession of DMT are prohibited. In the United Kingdom, "Fungus (of any kind) which contains psilocin or an ester of psilocin" is controlled as a Class A drug under the MDA and Schedule 1 drug under the MDR. As psilocybin is a phosphate ester of psilocin, even if it is isolated from psilocin, it will still be treated as a Class A drug under the MDA and as a Schedule 1 drug under the MDR. Schedule 1 drugs can only be lawfully manufactured, produced, possessed and supplied under a controlled drugs domestic licence issued by the UK Home Office.
There is no guarantee that psychedelic drugs or psychedelic inspired drugs will ever be approved as medicines in any jurisdiction in which the Corporation operates. All activities involving such substances by or on behalf of the Corporation are conducted in accordance with applicable federal, provincial, state and local laws. Further, all facilities engaged with such substances by or on behalf of the Corporation do so under current licences and permits issued by appropriate federal, provincial and local governmental agencies. While the Corporation is focused on programs using psychedelic inspired compounds, the Corporation does not have any direct or indirect involvement with the illegal selling, production or distribution of any substances in the jurisdictions in which it operates and does not intend to have any such involvement. However, the laws and regulations generally applicable to the industry in which the Corporation is involved in may change in ways currently unforeseen. Any amendment to or replacement of existing laws or regulations, including the classification or re-classification of the substances the Corporation is developing or working with, which are matters beyond the Corporation's control, may cause the Corporation's business, financial condition, results of operations and prospects to be adversely affected or may cause the Corporation to incur significant costs in complying with such changes or it may be unable to comply therewith. A violation of any applicable laws and regulations of the jurisdictions in which the Corporation operates could result in significant fines, penalties, administrative sanctions, convictions or settlements arising from civil proceedings initiated by either government entities in the jurisdictions in which the Corporation operates, or private citizens or criminal charges.
The loss of the necessary licences and permits for any of the above scheduled drugs could have an adverse effect on the Corporation's operations.
The psychedelic drug industry is a fairly new industry and the Corporation cannot predict the impact of the ever-evolving compliance regime in respect of this industry. Similarly, the Corporation cannot predict the time required to secure all appropriate regulatory approvals for future products, or the extent of testing and documentation that may, from time to time, be required by governmental authorities. The impact of compliance regimes, any delays in obtaining, or failure to obtain regulatory approvals may significantly delay or impact the development of markets, its business and products, and sales initiatives and could have a material adverse effect on the business, financial condition and operating results of the Corporation.
The success of the Corporation's business is dependent on the reform of controlled substances laws pertaining to psilocybin. If controlled substances laws are not favourably reformed in Canada, the United States, the Netherlands, the United Kingdom, and other global jurisdictions, the commercial opportunity that the Corporation is pursuing may be highly limited.
The Corporation makes no medical, treatment or health benefit claims about the Corporation's proposed products. The FDA, Health Canada, the EMA or other similar regulatory authorities have not evaluated claims regarding psilocybin, DMT, psilocybin analogues, or other psychedelic compounds. The efficacy of such products have not been confirmed by approved research. There is no assurance that the use of psilocybin, DMT, psilocybin analogues, or other psychedelic compounds can diagnose, treat, cure or prevent any disease or condition. Vigorous scientific research and clinical trials are needed. The Corporation has not conducted clinical trials for the use of its proposed products. Any references to quality, consistency, efficacy and safety of potential products do not imply that the Corporation verified such in clinical trials or that the Corporation will complete such trials. If the Corporation cannot obtain the approvals or research necessary to commercialize its business, it may have a material adverse effect on the Corporation's performance and operations.
"Foreign Private Issuer" Status Under the U.S. Securities Laws
The Corporation is a "foreign private issuer", under applicable U.S. federal securities laws, and is, therefore, not subject to the same requirements that are imposed upon U.S. domestic issuers by the SEC. Under the Exchange Act, the Corporation is subject to reporting obligations that, in certain respects, are less detailed and less frequent than those of U.S. domestic reporting companies. As a result, the Corporation does not file the same reports that a U.S. domestic issuer would file with the SEC, although the Corporation is required to file with or furnish to the SEC the continuous disclosure documents that it is required to file in Canada under Canadian securities laws. In addition, the Corporation's officers, directors, and principal shareholders are exempt from the reporting and short-swing profit recovery provisions of Section 16 of the Exchange Act. Therefore, the Corporation's shareholders may not know on as timely a basis when the Corporation's officers, directors and principal shareholders purchase or sell Common Shares, as the reporting periods under the corresponding Canadian insider reporting requirements are longer.
As a foreign private issuer, the Corporation is exempt from the rules and regulations under the Exchange Act related to the furnishing and content of proxy statements. The Corporation is also exempt from Regulation FD, which prohibits issuers from making selective disclosures of material non-public information. While the Corporation complies with the corresponding requirements relating to proxy statements and disclosure of material non-public information under Canadian securities laws, these requirements differ from those under the Exchange Act and Regulation FD and shareholders should not expect to receive the same information at the same time as such information is provided by U.S. domestic companies. In addition, the Corporation may not be required under the Exchange Act to file annual and quarterly reports with the SEC as promptly as U.S. domestic companies whose securities are registered under the Exchange Act.
Risks Related to an Offering
Speculative Nature of Investment Risk
An investment in the Common Shares and the Corporation's prospects generally are speculative due to the risky nature of its business and the present stage of its development. Investors may lose their entire investment. There is no assurance that risk management steps taken will avoid future loss due to the occurrence of the risks described or incorporated by reference herein, or other unforeseen risks. If any of such risks actually occur, then the Corporation's business, financial condition and operating results could be adversely affected. Investors should carefully consider all risks and consult with their professional advisors to assess any investment in the Corporation.
Negative Operating Cash Flow and Going Concern
The Corporation has had negative cash flow from operating activities since inception. Drug development involves long lead times, is very expensive and involves many variables of uncertainty. As such, significant capital investment will be required to achieve the Corporation's existing plans. The Corporation's net losses have had and will continue to have an adverse effect on, among other things, shareholder equity, total assets and working capital. The Corporation expects that losses may fluctuate from quarter to quarter and year to year, and that such fluctuations may be substantial based on the stage of development of its principal programs. The Corporation cannot predict when it will become profitable, if at all. Accordingly, the Corporation may be required to obtain additional financing in order to meet its future cash commitments.
The threat of the Corporation's ability to continue as a going concern will be removed only when, in the opinion of the Corporation's auditor, the Corporation's revenues have reached a level that is able to sustain its business operations. If the Corporation is unable to obtain additional financing from outside sources and eventually generate enough revenues, the Corporation may be forced to sell a portion or all of the Corporation's assets, or curtail or discontinue the Corporation's operations. If any of these events happen, shareholders could lose all or part of their investment. The Corporation's financial statements do not include any adjustments to the Corporation's recorded assets or liabilities that might be necessary if the Corporation becomes unable to continue as a going concern. See "Risk Factors - Risks Related to the Offering - Potential Need for Additional Financing".
Discretion over the Use of Proceeds
While detailed information regarding the use of proceeds from the sale of the Securities will be described in the applicable Prospectus Supplement, the Corporation will have broad discretion over the use of net proceeds from an offering by the Corporation of its Securities. There may be circumstances where, for sound business reasons, a reallocation of funds may be deemed prudent or necessary. In such circumstances, the net proceeds will be reallocated at the Corporation's sole discretion.
Management will have discretion concerning the use of proceeds ascribed in the applicable Prospectus Supplement as well as the timing of their expenditures. As a result, an investor will be relying on the judgment of management for the application of the proceeds. Management may use the net proceeds described in a Prospectus Supplement in ways that an investor may not consider desirable. The results and the effectiveness of the application of the proceeds are uncertain. If the proceeds are not applied effectively, the Corporation's results of operations may suffer. See "Use of Proceeds".
Need for Additional Financing
The continued development of the Corporation will require additional financing. The Corporation's activities do have scope for flexibility in terms of the amount and timing of expenditures, and expenditures may be adjusted accordingly. However, further operations will require additional capital and will depend on the Corporation's ability to obtain financing through debt, equity or other means. The Corporation's ability to meet its obligations and maintain operations may be contingent upon successful completion of additional financing arrangements. There is no assurance that the Corporation will be successful in obtaining the required financing in the future or that such financing will be available on terms acceptable to the Corporation. In addition, any future financing may also be dilutive to existing shareholders of the Corporation. See "Risk Factors - Risks Related to an Offering - Negative Operating Cash Flow and Going Concern" and "- Potential Dilution".
Volatile Market Price of Corporation's Common Shares
The securities market in Canada has recently experienced a high level of price and volume volatility, and the market prices of securities of many companies have experienced wide fluctuations in price which have not necessarily been related to the operating performance, underlying asset values or prospects of such companies. There can be no assurance that continual fluctuations in price will not occur. It may be anticipated that any market for the Common Shares will be subject to market trends generally, notwithstanding any potential success of the Corporation. The value of the Common Shares distributed hereunder will be affected by such volatility.
The volatility of the Common Shares may affect the ability of holders to sell the Common Shares at an advantageous price or at all. Market price fluctuations in the Common Shares may be adversely affected by a variety of factors relating to the Corporation's business, including fluctuations in the Corporation's operating and financial results, such results failing to meet the expectations of securities analysts or investors and downward revisions in securities analysis' estimates in connection therewith, sales of additional Common Shares, governmental regulatory action, adverse change in general market conditions or economic trends, acquisitions, dispositions or other material public announcements by the Corporation or its competitors, along with a variety of additional factors, including, without limitation, those set forth under the heading "Cautionary Note Regarding Forward-Looking Information". In addition, the market price for securities on stock markets, including the Neo is subject to significant price and trading fluctuations. These fluctuations have resulted in volatility in the market prices of securities that often has been unrelated or disproportionate to changes in operating performance. These broad market fluctuations may materially adversely affect the market price of the Corporation.
Additionally, the value of the Common Shares is subject to market value fluctuations based upon factors that influence the Corporation's operations, such as legislative or regulatory developments, competition, technological change and changes in interest rates or foreign exchange rates. There can be no assurance that the market price of the Common Shares will not experience significant fluctuations in the future, including fluctuations that are unrelated to the Corporation's performance. As at the date of this Prospectus, only the Common Shares are listed on a securities exchange and may be purchased in the secondary market.
Potential Dilution
The Corporation's articles of incorporation and by-laws allow it to issue an unlimited number of Common Shares for such consideration and on such terms and conditions as established by the board of directors of the Corporation, in many cases, without the approval of the Corporation's shareholders. The Corporation cannot predict the size of future issuances of Common Shares or other Securities or the effect that future issuances and sales of Common Shares or other Securities will have on the market price of the Corporation's Securities. Issuances of a substantial number of additional Securities, or the perception that such issuances could occur, may adversely affect prevailing market prices for the Common Shares. With any additional issuance of Common Shares, investors will suffer dilution to their voting power and the Corporation may experience dilution in its earnings per share. "Risk Factors - Risks Related to an Offering - Potential Need for Additional Financing".
Market for Securities
There is currently no market through which the Securities, other than the Common Shares, may be sold and, unless otherwise specified in the applicable Prospectus Supplement, such unlisted Securities may not be listed on any securities or stock exchange or any automated dealer quotation system. As a consequence, purchasers may not be able to resell such unlisted Securities purchased under this Prospectus. This may affect the pricing of the Corporation's Securities, other than the Common Shares, in the secondary market, the transparency and availability of trading prices, the liquidity of these Securities and the extent of issuer regulation. There can be no assurance that an active trading market for the Securities, other than the Common Shares, will develop or, if developed, that any such market, including for the Common Shares, will be sustained.
Enforcement of Civil Liabilities
Certain of the Corporation's subsidiaries and assets are located outside of Canada. Accordingly, it may be difficult for investors to enforce within Canada any judgments obtained against the Corporation, including judgments predicated upon the civil liability provisions of applicable Canadian securities laws or otherwise. Consequently, investors may be effectively prevented from pursuing remedies against the Corporation under Canadian securities laws or otherwise.
The Corporation has subsidiaries incorporated in the United States. It may not be possible for shareholders to effect service of process outside of Canada against the directors and officers of the Corporation who are not resident in Canada. In the event a judgment is obtained in a Canadian court against one or more of such persons for violations of Canadian securities laws or otherwise, it may not be possible to enforce such judgment against persons not resident in Canada. Additionally, it may be difficult for an investor, or any other person or entity, to assert Canadian securities law or other claims in original actions instituted in the United States. Courts in such jurisdiction may refuse to hear a claim based on a violation of Canadian securities laws or otherwise on the grounds that such jurisdiction is not the most appropriate forum to bring such a claim. Even if a foreign court agrees to hear a claim, it may determine that the local law, and not Canadian law, is applicable to the claim. If Canadian law is found to be applicable, the content of applicable Canadian law must be proven as a fact, which can be a time-consuming and costly process. Certain matters of procedure will also be governed by foreign law.
Most of the officers and directors named in this Prospectus are not residents of the United States, and some of the Corporation's assets and all or a substantial portion of the assets of such person are located outside of the United States.
The Corporation has been advised that, subject to certain limitations, a judgment of a United States court predicated solely upon civil liability under United States federal securities laws may be enforceable in Canada if the United States court in which the judgment was obtained has a basis for jurisdiction in the matter that would be recognized by a Canadian court for the same purposes. The Corporation has also been advised, however, that there is substantial doubt whether an action could be brought in Canada in the first instance on the basis of liability predicated solely upon United States federal securities laws or any such state securities or "blue sky" laws.
The Corporation is filing with the SEC, concurrently with the Registration Statement, an appointment of agent for service of process on Form F-X. Under the Form F-X, the Corporation appointed C T Corporation System as its agent for service of process in the United States in connection with any investigation or administrative proceeding conducted by the SEC, and any civil suit or action brought against or involving the Corporation in a United States court, arising out of or related to or concerning the offering of the Securities.
CERTAIN FEDERAL INCOME TAX CONSIDERATIONS
The applicable Prospectus Supplement will include a general summary of certain Canadian federal income tax consequences which may be applicable to a purchaser of Securities offered thereunder. The applicable Prospectus Supplement may also describe certain United States federal income tax consequences which may be applicable to a purchaser of Securities hereunder by an initial investor who is a United States person (within the meaning of the United States Internal Revenue Code of 1986, as amended). Investors should read the tax discussion in any Prospectus Supplement with respect to a particular offering and consult their own tax advisors with respect to their own particular circumstances.
LEGAL MATTERS
Certain legal matters in connection with the offering of the Securities will be passed upon by Aird & Berlis LLP on behalf of the Corporation. As at the date of this Prospectus, the designated professionals of Aird & Berlis LLP, as a group, beneficially own, directly or indirectly, less than one percent of the securities of the Corporation.
AUDITORS, TRANSFER AGENT AND REGISTRAR
Zeifmans LLP, Chartered Professional Accountants, are the auditors of the Corporation and have confirmed that they are independent of the Corporation within the meaning of the relevant rules and related interpretations prescribed by the relevant professional bodies in Canada and any applicable legislation or regulation. Neither Zeifmans LLP nor any designated professional thereof, had any registered or beneficial interest in any securities or other property of the Corporation at the time they prepared the relevant financial statements incorporated by reference in this Prospectus or at any time thereafter
The registrar and transfer agent of the Common Shares is Odyssey Trust Company at its principal office in Calgary, Alberta.
PART II
INFORMATION NOT REQUIRED TO BE DELIVERED TO
OFFEREES OR PURCHASERS
Indemnification of Directors and Officers
Under the Business Corporations Act (Ontario), the Registrant may indemnify a director or officer of the Registrant, a former director or officer of the Registrant or another individual who acts or acted at the Registrant's request as a director or officer, or an individual acting in a similar capacity, of another entity (each of the foregoing, an "individual"), against all costs, charges and expenses, including an amount paid to settle an action or satisfy a judgment, reasonably incurred by the individual in respect of any civil, criminal, administrative, investigative or other proceeding in which the individual is involved because of that association with the Registrant or other entity, on the condition that (i) such individual acted honestly and in good faith with a view to the best interests of the Registrant or, as the case may be, to the best interests of the other entity for which the individual acted as a director or officer or in a similar capacity at the Registrant's request; and (ii) if the matter is a criminal or administrative action or proceeding that is enforced by a monetary penalty, the Registrant shall not indemnify the individual unless the individual had reasonable grounds for believing that his or her conduct was lawful.
Further, the Registrant may, with the approval of a court, indemnify an individual in respect of an action by or on behalf of the Registrant or other entity to obtain a judgment in its favor, to which the individual is made a party because of the individual's association with the Registrant or other entity as a director or officer, a former director or officer, an individual who acts or acted at the Registrant's request as a director or officer, or an individual acting in a similar capacity, against all costs, charges and expenses reasonably incurred by the individual in connection with such action, if the individual fulfills the conditions in (i) and (ii) above. Such individuals are entitled to indemnification from the Registrant in respect of all costs, charges and expenses reasonably incurred by the individual in connection with the defense of any civil, criminal, administrative, investigative or other proceeding to which the individual is subject because of the individual's association with the Registrant or other entity as described above, provided the individual seeking an indemnity: (A) was not judged by a court or other competent authority to have committed any fault or omitted to do anything that the individual ought to have done; and (B) fulfills the conditions in (i) and (ii) above.
The by-laws of the Registrant provide that, subject to the Business Corporations Act (Ontario), the Registrant shall indemnify a director or officer of the Registrant, a former director or officer of the Registrant or another individual who acts or acted at the Registrant's request as a director or officer, or an individual acting in a similar capacity, or another entity, against all costs, charges and expenses, including an amount paid to settle an action or satisfy a judgment, reasonably incurred by the individual in respect of any civil, criminal, administrative, investigative or other proceeding in which the individual is involved because of that association with the Registrant or other entity, if: (i) the individual acted honestly and in good faith with a view to the best interests of the Registrant or, as the case may be, to the best interest of the other entity for which the individual acted as a director or officer or in a similar capacity at the Registrant's request and (ii) in the case of a criminal or administrative action or proceeding that is enforced by a monetary penalty, the person had reasonable grounds for believing that the individual's conduct was lawful.
The Registrant maintains directors' and officers' liability insurance which insures directors and officers for losses as a result of claims against the directors and officers of the Registrant in their capacity as directors and officers and also reimburses the Registrant for payments made pursuant to the indemnity provisions under the by-laws of the Registrant and the Business Corporations Act (Ontario).
* * *
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers or persons controlling the Registrant pursuant to the foregoing provisions, the Registrant has been informed that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act, and is therefore unenforceable.
EXHIBIT INDEX
Exhibit
Number |
|
Description |
| 4.1 |
|
The annual information form of the Registrant dated June 27, 2023 for the year ended March 31, 2023 (incorporated by reference from Exhibit 99.1 of the Registrant’s Annual Report on Form 40-F, filed with the Commission on June 27, 2023) |
| |
|
|
| 4.2 |
|
The audited consolidated financial statements of the Registrant and the notes thereto as at and for the fiscal year ended March 31, 2023, together with the auditor's report thereon (incorporated by reference from Exhibit 99.2 of the Registrant’s Annual Report on Form 40-F, filed with the Commission on June 27, 2023) |
| |
|
|
| 4.3 |
|
Management’s discussion and analysis of the Registrant for the year ended March 31, 2023 (incorporated by reference from Exhibit 99.3 of the Registrant’s Annual Report on Form 40-F, filed with the Commission on June 27, 2023) |
| |
|
|
| 4.4 |
|
The unaudited interim condensed consolidated financial statements of the Registrant and the notes thereto for the three months ended June 30, 2023 (incorporated by reference from Exhibit 99.1 of the Registrant’s Form 6-K, furnished to the Commission on August 14, 2023) |
| |
|
|
| 4.5 |
|
Management’s discussion and analysis of the Registrant for the three months ended June 30, 2023 (incorporated by reference from Exhibit 99.2 of the Registrant’s Form 6-K, furnished to the Commission on August 14, 2023) |
| |
|
|
| 4.6 |
|
Management information circular of the Registrant dated July 13, 2022 relating to an annual meeting of shareholders of the Registrant held on August 15, 2022 (incorporated by reference from Exhibit 99.1 of the Registrant’s Form 6-K, furnished to the Commission on July 25, 2022) |
| |
|
|
| 4.7* |
|
The material change report of the Registrant dated May 31, 2023 (incorporated by reference from Exhibit 99.1 of the Registrant’s Form 6-K, furnished to the Commission on June 1, 2023) |
| |
|
|
| 4.8 |
|
The material change report of the Registrant dated August 8, 2023 (incorporated by reference from Exhibit 99.1 of the Registrant’s Form 6-K, furnished to the Commission on August 9, 2023) |
| |
|
|
| 5.1 |
|
Consent of Zeifmans LLP |
| |
|
|
| 6.1* |
|
Powers of Attorney (included on the signature page of the Form F-10 Registration Statement filed with the Commission on June 16, 2023) |
| |
|
|
| 7.1* |
|
Form of Debt Indenture |
| |
|
|
| 107 |
|
Filing Fee Table |
| |
|
|
| |
|
*Previously Filed |
PART III
UNDERTAKING AND CONSENT TO SERVICE OF PROCESS
Item 1. Undertaking
The Registrant undertakes to make available, in person or by telephone, representatives to respond to inquiries made by the Commission staff, and to furnish promptly, when requested to do so by the Commission staff, information relating to the securities registered pursuant to Form F-10 or to transactions in said securities.
Item 2. Consent to Service of Process
A written Appointment of Agent for Service of Process and Undertaking on Form F-X for the Registrant and its agent for service of process has been filed.
Any change to the name or address of the agent for service of process of the Registrant shall be communicated promptly to the Commission by amendment to Form F-X referencing the file number of this Registration Statement on Form F-10.
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, as amended, the registrant certifies that it has reasonable grounds to believe that it meets all of the requirements for filing on Form F-10 and has duly caused this Amendment No. 1 to Registration Statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Toronto, Country of Canada on August 17, 2023.
| |
CYBIN INC. |
| |
|
| |
By: |
/s/ Greg Cavers |
| |
|
Name: |
Greg Cavers |
| |
|
Title: |
Chief Financial Officer |
Pursuant to the requirements of the Securities Act of 1933, this Registration Statement has been signed below by the following persons in the capacities indicated and on the dates indicated.
| Signature |
|
Capacity |
|
Date |
| |
|
|
|
|
| * |
|
Chief Executive Officer |
|
August 17, 2023 |
| Douglas Drysdale |
|
|
|
|
| |
|
|
|
|
| /s/ Greg Cavers |
|
|
|
|
| Greg Cavers |
|
Chief Financial Officer |
|
August 17, 2023 |
| |
|
|
|
|
| * |
|
|
|
|
| Eric So |
|
Director and Executive Chairman and President |
|
August 17, 2023 |
| |
|
|
|
|
| * |
|
|
|
|
| Eric Hoskins |
|
Director |
|
August 17, 2023 |
| |
|
|
|
|
| * |
|
|
|
|
| Grant Froese |
|
Director |
|
August 17, 2023 |
| |
|
|
|
|
| * |
|
|
|
|
| Mark Lawson |
|
Director |
|
August 17, 2023 |
| |
|
|
|
|
| * |
|
|
|
|
| Theresa Firestone |
|
Director |
|
August 17, 2023 |
| |
|
|
|
|
| * |
|
|
|
|
| Paul Glavine |
|
Director |
|
August 17, 2023 |
| *By: |
/s/ Greg Cavers |
| |
Name: |
Greg Cavers |
| |
Title |
Attorney-in-fact |
AUTHORIZED REPRESENTATIVE
Pursuant to the requirements of Section 6(a) of the Securities Act of 1933, as amended, the undersigned has signed this Amendment No. 1 to Registration Statement, in the capacity of the duly authorized representative of the Registrant in the United States, on August 17, 2023.
| |
/s/ Douglas Drysdale |
| |
Name: Douglas Drysdale |
| |
Title: Chief Executive Officer |

We consent to the incorporation by reference into the Amendment No. 1 to Registration Statement on Form F-10 (the "Form F-10/A") of Cybin Inc. (the "Company") being filed with the United States Securities and Exchange Commission, and any amendments thereto, of our report, dated June 27, 2023, on the consolidated statements of financial position of the Company as of March 31, 2023 and 2022, and the consolidated statements of loss and comprehensive loss, changes in shareholders' equity and cash flows for the years then ended, and notes to the consolidated financial statements, including a summary of significant accounting policies. We also consent to the reference to us under the caption "Auditors, Transfer Agent and Registrar" in the short-form base shelf prospectus contained in the Form F-10/A.
| |
/s/ Zeifmans LLP |
Toronto, Canada
August 17, 2023 |
Chartered Professional Accountants
Licensed Public Accountants |
| 201 Bridgeland Avenue | Toronto Ontario | M6A 1Y7 | Canada |
zeifmans.ca
T: 416.256.4000 |
|
 |
EX-FILING FEES
Calculation of Filing Fee Tables
FORM F-10
(Form Type)
CYBIN INC.
(Exact Name of Registrant as Specified in its Charter)
Table 1: Newly Registered and Carry Forward Securities
| |
Security Type |
Security
Class Title |
Fee
Calculation
or Carry
Forward
Rule |
|
Amount
Registered(1) |
|
|
Proposed
Maximum
Offering
Price
Per Unit |
|
|
Maximum
Aggregate
Offering Price(1)(3) |
|
|
Fee Rate |
|
|
Amount of
Registration Fee |
|
| Newly Registered Securities |
|
| Fees to be Paid |
Unallocated
(Universal) Shelf |
Common Shares, Warrants, Units, Debt Securities
Subscription Receipts |
Rule 457(o) |
$ |
24,855,731 |
(2)
|
|
(1) |
|
$ |
24,855,731 |
(2) |
$ |
0.0001102 |
|
$ |
2,740 |
(2)
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Fees Previously Paid |
Unallocated
(Universal) Shelf |
Common Shares, Warrants, Units, Debt Securities
Subscription Receipts |
Rule 457(o) |
$ |
94,006,167 |
|
|
(1) |
|
$ |
94,006,167 |
|
$ |
0.0001102 |
|
$ |
10,359 |
|
| |
Total Offering Amounts |
|
|
|
|
$ |
118,861,898 |
|
|
|
|
$ |
13,099 |
|
| |
Total Fees Previously Paid |
|
|
|
|
|
|
|
|
|
|
$ |
7,297 |
|
| |
Total Fee Offsets |
|
|
|
|
|
|
|
|
|
|
$ |
1,399 |
|
| |
Net Fee Due |
|
|
|
|
|
|
|
|
|
|
$ |
4,403 |
|
(1) There are being registered under this Registration Statement such indeterminate number of common shares, warrants, units, debt securities and subscription receipts of the Registrant, and a combination of such securities, separately or as units, as may be sold by the registrant from time to time, which collectively shall have an aggregate initial offering price not to exceed $118,861,898 (converted from CAD$160,000,000 at an exchange rate of CAD$1.00 = $1.3461, which was the daily average exchange rate reported by the Bank of Canada on August 14, 2023). The securities registered hereunder also include such indeterminate number of each class of identified securities as may be issued upon conversion, exercise or exchange of any other securities that provide for such conversion into, exercise for or exchange into such securities. Separate consideration may or may not be received for securities that are issuable on exercise, conversion or exchange of other securities. In addition, pursuant to Rule 416 under the Securities Act of 1933, as amended (the "Securities Act"), the common shares being registered hereunder include such indeterminate number of common shares as may be issuable with respect to the common shares being registered hereunder as a result of stock splits, stock dividends, distributions or similar transactions. The proposed maximum initial offering price per security will be determined, from time to time, by the registrant in connection with the sale of the securities under this Registration Statement.
(2) The Registrant previously paid $7,297 in registration fees (which included a $3,062 fee offset as described in Table 2 in Exhibit 107 of the Form F-10 filed June 16, 2023) in connection with the initial filing of this Registration Statement, which had an aggregate initial offering amount of $94,006,167 (converted from CAD$125,000,000 at an Exchange Rate of CAD$1.00 = $1.3297, which was the daily average exchange rate reported by the Bank of Canada on June 14, 2023). Pursuant to this Amendment No. 1 to the Registration Statement, the Registrant has increased the aggregate offering amount by CAD$35,000,000, or $24,855,731. Accordingly, the Registrant is paying an additional $2,740 in registration fees in connection with filing this Amendment No. 1 to the Registration Statement.
(3) Estimated solely for the purpose of calculating the amount of the registration fee pursuant to Rule 457(o) of the Securities Act.
Table 2: Fee Offset Claims and Sources
| |
Registrant or
Filer Name |
Form or Filing
Type |
File Number |
Initial Filing
Date |
Filing Date |
|
Fee Offset
Claimed |
|
|
Security Type
Associated
with Fee
Offset
Claimed |
|
|
Security
Title
Associated
with Fee
Offset
Claimed |
|
|
Unsold
Securities
Associated
with Fee Offset
Claimed |
|
|
Unsold
Aggregate
Offering Amount
Associated with
Fee Offset
Claimed |
|
|
Fee Paid with
Fee Offset
Source |
|
| |
Rule 457(p) |
|
| Fees Offset Claims |
Cybin Inc. |
F-10 |
333-259994(3) |
10/01/2021 |
|
$ |
1,399 |
|
|
Unallocated (Universal) Shelf |
|
|
Unallocated (Universal) Shelf |
|
|
Unallocated (Universal) Shelf |
|
$ |
15,089,589 |
|
|
|
|
| Fees Offset Sources |
Cybin Inc. |
F-10 |
333-259994(3) |
|
10/01/2021 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
$ |
1,399 |
|
(4) The Registrant previously paid $9,154.13 in registration fees with respect to the Registration Statement on Form F-10 (333-259994) on October 1, 2021 (the "Prior Registration Statement"), of which (i) $3,245 was utilized in connection with the Registrant's prospectus supplement filed August 8, 2022 to the Prior Registration Statement pursuant to which the Registrant offered common shares having an aggregate offering price of up to $35,000,000, (ii) $2,846 was utilized in connection with the Registrant's prospectus supplement filed May 31, 2023 to the Prior Registration Statement pursuant to which the Registrant (A) offered common shares having an aggregate offering price of up to $30,000,000 and (B) issued 2,538,844 common shares, the filing fee associated with such common shares was calculated in accordance with Rule 457(c) using the high and low sale prices of the common shares on the NYSE American Exchange on May 25, 2023, which is a date within five business days of filing such prospectus supplement, (iii) $765 was utilized in connection with the Registrant's prospectus supplement filed August 2, 2023 to the Prior Registration Statement pursuant to which the Registrant offered units having an aggregate offering price of $8,250,000 (the "Unit Offering") and (iv) $900 was utilized in connection with the Registrant's prospectus supplement filed August 4, 2023 to the Prior Registration Statement pursuant to which the Registrant registered 24,264,706 common shares issuable upon the exercise of 24,264,706 common share purchase warrants (the "Warrants") issued pursuant to the Unit Offering at an exercise price of $0.40 per Warrant (the "Exercise Price"), the filing fee associated with such common shares was calculated in accordance with Rule 457(g) using the Exercise Price. Therefore, $1,399 of the previously paid fees attributable to $15,089,589 of unsold securities that were previously registered under the Prior Registration Statement may be applied to the filing fees payable pursuant to this Registration Statement, and the Prior Registration Statement and the offering of the unsold securities registered under the Prior Registration Statement will be deemed terminated as of the effective date of this Registration Statement.
Cybin (AMEX:CYBN)
Graphique Historique de l'Action
De Avr 2024 à Mai 2024

Cybin (AMEX:CYBN)
Graphique Historique de l'Action
De Mai 2023 à Mai 2024
